MIELOPROLIFERATIVE DISORDERS INSTITUTUL REGIONAL DE ONCOLOGIE IASI CLINICA

 MPD are clonal diseases originating in pluripotential haematopoietic stem cell.")
 Neoplastic (clonal) disorders of hemopoietic stem cells Over-production of all")



 Increased number of Red cells Granulocytes Platelets (Note: myeloproliferation in")



 Polycythemia Vera Essential Thrombocythemia Myelofibrosis –")

")






 gene on chromosome 22 fused to the ABL (Ableson")

 is the fusion")

 Disease is biphasic, sometimes triphasic The clinical manifestations of CML")
 Features of anaemia – Pallor, dyspnoea, tachycardia Abnormal platelet function")
 Splenomegaly (70– 85%) - is the most common physical finding")
 Fever, purpura. Physical findings of leukostasis and hyperviscosity can occur")







 Complications – anemia,")


 Polymerase chain")
 or")
 in CML: 1) Allows")



/ 0(t) = Exp 0, 0116 (Age - 43,")


 – allogeneic transplantation")

")



 v.")

 = a 3 log reduction in BCR-ABL/BCR")




 “For the moment allogeneic bone marrow or stem")


 (Polycythaemia rubra vera) Definition of polycythemia Raised packed cell volume (PCV")

 (1) I. Absolute erythrocytosis (Polycythemia): A. Secondary erythrocytosis (abnormal increase of serum")
 (2) I. Absolute erythrocytosis (Polycythemia): A. Secondary erythrocytosis (abnormal increase of serum")
 Polycythaemia vera is a clonal stem cell disorder characterised by increased")
 Clinical features Age – 55 -60 years – May occur in")






 Criteria for PV Minor")






")

 ET is a clonal myeloproliferative disorder characterized by bone marrow hyperplasia")
 Neoplastic stem cell disorder causing dysregulated production of large numbers of")

 Numerous thrombocyte aggregates in peripheral")




, young (<")



 hyperproliferation of")
 – severe fatigue")




 Score Median Survival")
 in anemia from decreased red cell production")



- Slides: 110
MIELOPROLIFERATIVE DISORDERS INSTITUTUL REGIONAL DE ONCOLOGIE IASI CLINICA HEMATOLOGIE 2012 -2013
CHRONIC MYELOPROLIFERATIVE DISORDERS (MPD) MPD are clonal diseases originating in pluripotential haematopoietic stem cell. The clonal expansion results in increased and abnormal haematopoiesis and produces a group of interrelated syndromes, classified according to the predominant phenotypic expression of the myeloproliferative clone.
CHRONIC MYELOPROLIFERATIVE DISORDERS (MPD) Neoplastic (clonal) disorders of hemopoietic stem cells Over-production of all cell lines, with usually one line in particular Fibrosis is a secondary event Acute Myeloid Leukemia may occur
HAEMATOPOIESIS
HEMATOPOIETIC PROGENITORS Genetic Mutation National Cancer Institute
GENERALITIES Hemopoietic stem cell disorder – Clonal – Characterized by proliferation – Granulocytic – Erythroid – Megakaryocytic Interrelationship between – Polycythaemia – Essential thrombocythaemia – myelofibrosis
GENERALITIES Normal maturation (effective) Increased number of Red cells Granulocytes Platelets (Note: myeloproliferation in myelodysplastic syndrome is ineffective) Frequent overlap of the clinical, laboratory & morphologic findings Leucocytosis, thrombocytosis, increased megakaeryocytes, fibrosis & organomegaly blurs the boundaries Hepatosplenomegaly Sequestration of excess blood Extramedullary haematopoiesis Leukaemic infiltration
Rationale for classification Classification is based on the lineage of the predominant proliferation Level of marrow fibrosis Clinical and laboratory data (FBP, BM, cytogenetic & molecular genetic)
CHRONIC MYELOPROLIFERATIVE DISORDERS WHO Classification of CMPD • • Ch Myeloid leukemia Ch Neutrophillic leukemia Ch Eosinophillic leukemia / Hyper Eo Synd Polycythemia Vera Essential Thrombocythemia Myelofibrosis CMPD unclassifiable
MYELOPROLIFERATIVE DISORDERS MPD • PRV • ET • MF CML CMML MDS • RARS • RAEB II AML
CHRONIC MYELOPROLIFERATIVE DISORDERS Chronic Myeloid leukemia (BCR-ABL positive) Polycythemia Vera Essential Thrombocythemia Myelofibrosis – Specific clincopathologic criteria for diagnosis and distinct diseases, have common features – Increased number of one or more myeloid cells – Hepatosplenomegaly – Hypercatabolism – Clonal marrow hyperplasia without dysplasia – Predisposition to evolve
Bone marrow stem cell Clonal abnormality Granulocyte precursors Chronic myeloid leukemia Red cell precursors Polycythaemia rubra vera (PRV) 10% 70% AML Megakaryocytes Reactive fibrosis Essential thrombocytosis (ET) Myelofibrosis 10% 30%
CHRONIC MYELOGENOUS LEUKEMIA (CML)
CML - INTRODUCTION Clonal malignant myeloproliferative disorder characterized by increased proliferation of the granulocytic cell line without the loss of their capacity to differentiate Results in increases in myeloid cells, erythroid cells and platelets in peripheral blood and marked myeloid hyperplasia in the bone marrow Originate in a single abnormal haemopoietic stem cell
CML - BACKGROUND CML was the first human malignancy to be associated with a specific genetic lesion, the Philadelphia chromosome, harboring the BCRABL oncogene. Since then, it has become a paradigm for the discovery of molecular mechanisms and targeted therapeutic approaches in the field of hematologic neoplasias.
CML - EPIDEMIOLOGY CML accounts for 20% of all leukemias affecting adults. Frequency - 10 -15 new cases, each year, for one million population. In general, this disease occurs in the fourth and fifth decades of life. Younger patients aged 20 -29 years may be affected and may present with a more aggressive form, such as in accelerated phase or blast crisis. Sex ratio is 1, 4 -2, 2 (m/f).
CML - ETIOLOGY Etiology – Not clear – Little evidence of genetic factors linked to the disease – Increased incidence Survivors of the atomic disasters at Nagasaki & Hiroshima Post radiation therapy
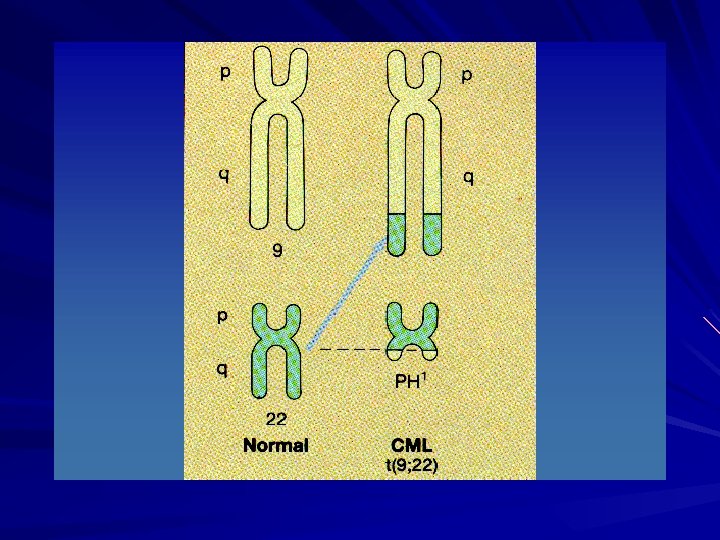
LEUKEMOGENESIS Philadelphia chromosome is an acquired cytogenetic anomaly that is characterizes in all leukaemic cells in CML 90 -95% of CML pts have Ph chromosome Reciprocal translocation of chromosome 22 and chromosome 9
CML - Ph CHROMOSOME
LEUKEMOGENESIS BCR (breakpoint cluster region) gene on chromosome 22 fused to the ABL (Ableson leukemia virus) gene on chromosome 9 Ph chromosome is found on myeloid, monocytic, erythroid, megakaryocytic, B-cells and sometimes T-cell proof that CML derived from pluripotent stem cell
LMC - CROMOZOMUL Ph
Consequences of p 210 BCR-ABL Molecular consequence of the t(9; 22) is the fusion protein BCR–ABL, which has increased in tyrosine kinase activity BCR-ABL protein transform hematopoietic cells so that their growth and survival become independent of cytokines It protects hematopoietic cells from programmed cell death (apoptosis) – – – deregulated cellular proliferation, decreased adherence of leukemia cells to the bone marrow stroma reduced apoptotic response to mutagenic stimuli
LMC - CROMOZOMUL Ph
CML – HISTORY (I) Disease is biphasic, sometimes triphasic The clinical manifestations of CML are insidious and are often discovered incidentally - 40% asymptomatic Chronic phase Splenomegaly often massive Symptoms related to hypermetabolism – – Weight loss Anorexia Lassitude Night sweats
CML – HISTORY (II) Features of anaemia – Pallor, dyspnoea, tachycardia Abnormal platelet function – Bruising, epistaxis, menorrhagia Hyperleukocytosis – – thrombosis Increased purine breakdown : gout Visual disturbances Priapism Some patients may present with complications – spleen infarction, spleen fracture, peptic ulcer with hemorrhagia.
CML – PHYSICAL (I) Splenomegaly (70– 85%) - is the most common physical finding in patients with CML. – The size of the spleen correlates with the peripheral blood granulocyte counts – A very large spleen is usually a harbinger of the transformation into an acute blast crisis form of the disease Hepatomegaly also occurs, although less commonly than splenomegaly , in 20– 45%, Lymphadehopaties - signifie the evolution to the accelerated/blastic phase.
CML – PHYSICAL (II) Fever, purpura. Physical findings of leukostasis and hyperviscosity can occur in some patients, with extraordinary elevation of their WBC counts, exceeding 300, 000 -600, 000 cells/mmc : – headache, – dizziness, – vertigo, – tinnitus, – visual disturbances, – angina pectoris, or – intermittent claudications.
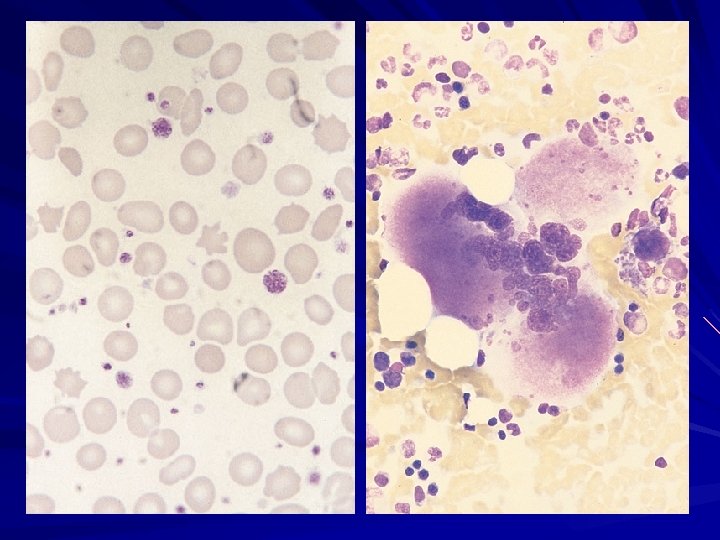
CML – PERIPHERAL BLOOD Leucocytosis (a total WBC count of 20, 000 -over 100. 000 cells/m. L) with circulating immature cells from the bone marrow, such as myeloblasts, myelocytes, metamyelocytes, and nucleated red blood cells, mimicking the findings in the bone marrow. Basophilia - 2 -4%, over 7% in 10 – 15 % patients Eosinophilia Mild-to-moderate anemia usually normochromic and normocytic Thrombocitosis - 500. 000– 600. 000/mm 3, to 1– 2 million/mm 3.
CML – PERIPHERAL BLOOD
CML – PERIPHERAL BLOOD
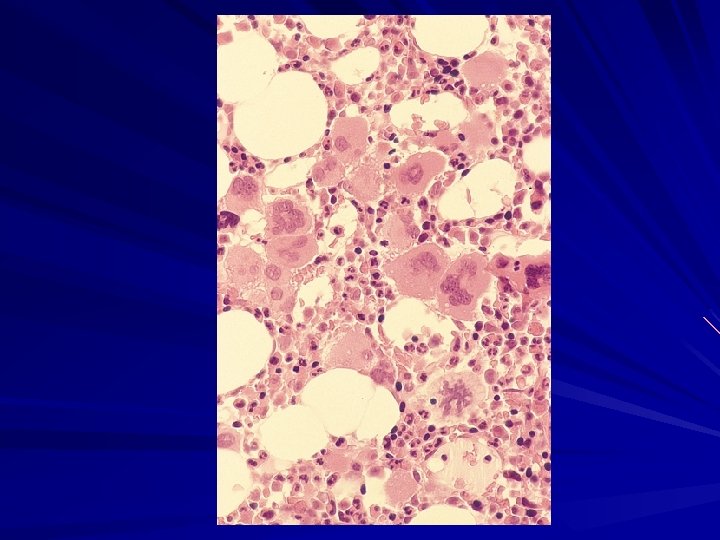
CML – BONE MARROW Marrow aspirate - Bone marrow is – – – Hypercellular (reduced fat spaces) Myeloid: erythroid ratio – 10: 1 to 30: 1 (N : 2: 1) Myelocyte predominant cell, blasts less 10% Megakaryocytes increased & dysplastic Increase reticulin fibrosis in 30 -40% BOM - confirms the hyperplasia of hematopoietic tissue. Mild fibrosis is often seen in the reticulin stain.
CML – BONE MARROW
CML – BONE MARROW
CML – BIOLOGY the leukocyte alkaline phosphatase stains very low to absent in most cells, resulting in a low score. markedly elevated serum vitamin B-12–binding protein (TC-I); hemostasis thrombocytopathia with prolonged bleeding time and decreased platelet aggregability. hyperuricemia and hyperuricuria, elevated LDH; hyperhistaminemia;
CML - EVOLUTION Chronic phase Accelerated phase Acute phase (blastic phase) Complications – anemia, infections, – bleeding or thrombosis (priapism), – spleen infarction or fracture, – pulmonary (infarction, infections), – bone (pains, distruction, hypercalcemia), – neurologic (leucostasis, hemorragia, thrombosis), – metabolic (gout)
LMC – ACCELERATED PHASE Median duration is 3. 5 – 5 yrs before evolving to more aggressive phases May last for several months. – Clinical features Increasing splenomegaly refractory to chemo Increasing chemotherapy requirement – Lab features Blasts>15% in blood Blast & promyelocyte > 30% in blood Basophil 20% in blood Thrombocytopenia Cytogenetic: clonal evolution
CML – BLASTIC PHASE Criteria : – – – Resembles acute leukaemia Diagnosis requires > 30% blast in marrow 2/3 transform to myeloid blastic phase and 1/3 to lymphoid blastic phase – Survival : 9 mos vs 3 mos (lym vs myeloid)
BCR-ABL – detection techniques Conventional cytogenetic analysis Fluorescece in situu hybridisation (FISH) Polymerase chain reaction (PCR) Reverse transcriptase – PCR (RT-PCR) Real-time quantitative PCR (RQ-PCR) for BCRABL m. RNA Utility : – Diagnosis positif and differential – Monitorising drug therapy – Assesment of minimal residual disease (MRD)
Diagnostic Considerations in Chronic Myeloid Leukemia Demonstrating the presence of the t(9; 22) or its gene product is absolutely essential in diagnosing a patient with CML Karyotyping in CML 1) Allows for the diagnosis of CML 2) Requires a bone marrow aspirate for optimal metaphases 3) Allows for evaluation of clonal evolution as well as additional chromosomal abnormalities in the non. Ph+ clones 4) Occasional cryptic and complex karyotypes can result in the missed identification of the t(9; 22) Source Undetermined
Diagnostic Considerations in Chronic Myeloid Leukemia Fluorescence in-situ hybridization (FISH) in CML: 1) Allows for the diagnosis of CML 2) Does not require a bone marrow aspirate for optimal results 3) Allows for the identification of potential duplications of the Ph chromosome 4) Allows for the identification of the loss of the der (9) chromsome 5) Allows for the identification of cryptic translocations involving Bcr-Abl Bcr- Ch 22 Abl – Ch 9 Bcr-Abl Fusion Source Undetermined
LMC - FISH
Diagnostic Considerations in Chronic Myeloid Leukemia Bcr-Abl c. DNA Quantitative RT-PCR for Bcr-Abl in CML Bcr 1) Allows for the diagnosis of CML Abl 2) Does not require a bone marrow aspirate for optimal results 3) Can quantify the amount of disease 4) Allows for the identification of cryptic translocations involving Bcr-Abl 5) Many primers sets only detect the p 190 and/or the p 210 translocation and may miss the p 230 or alternative translocations Source Undetermined
CML – PROGNOSTIC Poor prognosis features in patients with CML include : – – – – older age symptomatic presentation poor performance status Hepatomegaly, splenomegaly negative Ph chromosome or bcr-abl anemia thrombocytopenia/thrombocytosis, basophilia, or myelofibrosis (increased reticulin or collagen) – longer time to hematologic remission with myelosuppression therapy
CML – PROGNOSTIC Sokal Score (+)/ 0(t) = Exp 0, 0116 (Age - 43, 4) + 0, 0345 (spleen - 7, 51)+ 0, 188 (Pt/700)2 0, 563 + 0, 0887 (blasts - 2, 10) Score – IS = < 0, 8 – IS = 0, 8 - 1, 2 – IS = >1, 2 Median survival Survival at 48 months 60 months 44 months 32 months 62% 43% 33%
CML – GENERAL MANAGEMENT Discussion with family – The disease & diagnosis – Prognosis – Choices of treatment Cytotoxic drug vs bone marrow transplant Side effect
CML – PRINCIPLES OF TREATMENT Relieve symptoms of hyperleukocytosis, splenomegaly and thrombocytosis – Hydration – Chemotherapy (bulsuphan, Hydoxyurea) Control and prolong chronic phase (non-curative) – – – alpha interferon+chemotherapy imatinib mesylate chemotherapy (hydroxyurea)
CML – PRINCIPLES OF TREATMENT Treatment cont… Eradicate malignant clone (curative) – allogeneic transplantation – alpha interferon ? – imatinib mesylate/STI 571 ? (Thyrosine kinase inhibitor)
LMC – MEDICAL CARE Myelosupresive drugs Interferon Imatinib Allogenic hematopoietic stem cell transplantation Autologous hematopoietic stem cell transplantation Experimental drugs
CHEMOTHERAPY Busulphan – Alkylating agent – Preferred in older pts (not candidate for transplant) – Side effect : prolonged myelosuppression Pulmonary fibrosis Skin pigmentation infertility
CHEMOTHERAPY Hydoxyures – Fewer side effect – Acts by inhibiting the enzyme ribonucleotide reductase Haematological remissions obtain in 80% for both drugs However disease progression not altered and persistence of Ph chromosome containing clone
CHEMOTHERAPY Recombinant human α- Interferon – Optimal dose : 3 -5 MUI/m 2/d – Prolong chronic phase and increase survival – Haematogical and cytogenetic remission – Side effect Flu like symptoms Fever and chills Anorexia Depression
CML – THERAPEUTICAL AIM Therapeutic response – Hematologic response – Cytogenetic response – Molecular response Survival – Overall survival (OS) – Progression-free survival (PFS) – Disease free survival (DFS) – Time to progression (TTP)
HEMATOLOGIC RESPONSE Complete hematologic response v. WBC count normalised (under 10. 000/mm 3) v. Disapearance of immature cells of peripheral blood v. Platelets count normalised (under 350. 000/mm 3) v. Disapearance of all signs and symptoms of disease v. Spleen normalised Partial hematologic response v. Decreasing with at least 50% of leucocytosis (10. 000 si 20. 000/mm 3 ) vor complete hematologic response with persistent splenomegaly
CYTOGENETIC RESPONSE % cells Ph+ in bone marrow Complete : Partial : Major : Minimal : Eşec : 0 < 35 complet + parţial 35 - 95 > 95 100
MOLECULAR RESPONSE Major molecular response (Maj. MR) = a 3 log reduction in BCR-ABL/BCR level when compared to the median pretreatment level. Major molecular response(Maj. MR) = BCR-ABL/BCR 0. 045% Complete molecular response(CMR) = BCR-ABL unedetectable or reduction BCR -ABL/BCR with > 4. 5 log
Normal Bcr-Abl Signaling* The kinase domain activates a substrate protein, eg, PI 3 kinase, by phosphorylation This activated substrate initiates a signaling cascade culminating in cell proliferation and survival Substrate Effector Bcr-Abl ADP P PPP ATP PPP ADP = adenosine diphosphate; ATP = adenosine triphosphate; P = phosphate. Savage and Antman. N Engl J Med. 2002; 346: 683 Scheijen and Griffin. Oncogene. 2002; 21: 3314. SIGNALING
Imatinib Mesylate: Mechanism of Action* Imatinib mesylate occupies the ATP binding pocket of the Abl kinase domain This prevents substrate phosphorylation and signaling A lack of signaling inhibits proliferation and survival Savage and Antman. N Engl J Med. 2002; 346: 683. Bcr-Abl P ATP PPP Imatinib mesylate SIGNALING
Imatinib Mesylate in Chronic Phase CML Following IFN- Failure: Overall Survival* 1. 0 Survival probability 0. 9 0. 8 0. 7 0. 6 0. 5 0. 4 Total 261 251 0. 3 0. 2 Dead 31 193 0. 1 Imatinib mesylate Others (P<0. 0001) 0 0 24 48 72 96 Months Kantarjian et al. Blood. 2004; 1979. Copyright American Society of Hematology, used with permission.
Imatinib - schema terapeutica Recomendede dose : – 400 mg/d for patients in chronic phase – 600 – 800 mg/d for patients in accelerated phase or in blastic phase. The daily dose is administred in one dose, with lunch, associated withat least 250 ml water
Allogenic hematopoietic stem cell transplantation (AHSCT) “For the moment allogeneic bone marrow or stem cell transplantation is the best and only one treatment for cure of this disease. ” But The procedure is limited by the existance of a potential donor and the high toxicity which limits the age of the patients with potential indication for transplantation.
Source of HSC Source : Bone marrow – bone marrow transplantation Peripheral blood – peripheral blood HSC transplantation Peripheral blood source is preferred - because Quicker engraftment Less regimen related toxicity Shorter post-therapeutic aplasia Longer disease free – Gv. L effect is stronger
POLYCYTHEMIA VERA
POLYCYTHEMIA VERA (PV) (Polycythaemia rubra vera) Definition of polycythemia Raised packed cell volume (PCV / HCT) Male > 0. 51 (50%) Female > 0. 48 (48%) Classification Absolute Primary proliferative polycythaemia (polycythaemia vera) Secondary polycythaemia Idiopathic erythrocytosis Apparent Plasma volume or red cell mass changes
POLYCYTHEMIA True / Absolute – Primary Polycythemia – Secondary Polycythemia Epo dependent – Hypoxia independent Epo independent Apparent / Relative – Reduction in plasma volume
ERYTHROCYTOSIS (Classification) (1) I. Absolute erythrocytosis (Polycythemia): A. Secondary erythrocytosis (abnormal increase of serum erythropoietin level) 1. Erythrocytosis secondary to decreased tissue oxygenation: a) chronic lung diseases b) cyanotic congenital heart diseases c) high-altitude erythrocytosis (Monge disease) d) hypoventilation syndromes (Sleep apnoe) e) hemoglobin-oxygen dissociation abnormalities - hemoglobinopathies associated with high oxygen affinity - carboxyhemoglobin in „smoker’s polycythemia” 67
ERYTHROCYTOSIS (Classification) (2) I. Absolute erythrocytosis (Polycythemia): A. Secondary erythrocytosis (abnormal increase of serum erythropoietin level) 2. Secondary to aberrant erythropoietin production or response: a) Erythropoietin-producting tumors: hepatoma, uterine leiomyoma, cerebellar hemangioblastoma, ovarian carcinoma, pheochromocytoma b) Renal diseases: renal cell carcinoma, kidney cysts and hydronephrosis, renal transplantation. c) Androgen abuse: adrenal cortical hypersecretion, exogenous androgens B. Primery erythrocytosis 1. Polycythemia vera 2. Familial erythrocytosis II. Relative erythrocytosis (pseudopolycythemia): 1. Hemoconcentration 2. Spurious polycythemia (Gaisboek syndrome) 68
POLYCYTHEMIA VERA (PV) Polycythaemia vera is a clonal stem cell disorder characterised by increased red cell production Abnormal clones behave autonomous Same abnormal stem cell give rise to granulocytes and platelets Disease phase Proliferative phase “Spent” post-polycythaemic phase Rarely transformed into acute leukemia Epidemiology – 2 -3 / 100000 – Median age at presentation: 55 -60 – M/F: 0. 8: 1. 2
POLYCYTHEMIA VERA (PV) Clinical features Age – 55 -60 years – May occur in young adults and rare in childhood Symptoms common to all erythrocytosis – Headache, mental acuity, weakness Symptoms more specific to P vera and myeloproliferative diseases. – – – Pruritis after bathing Erythromelalgia Hypermetabolic symptoms Thrombosis (arterial or venous) Hemorrhage
POLYCYTHEMIA VERA physical examination Splenomegaly – is present in 75% of patients at the time of diagnosis. 2. Hepatomegaly - is present in approximately 30% of patients at the time of diagnosis. 3. Hypertension 4. On examination of the eye grounds, the vessels may be engorged, tortuous, and irregular in diameter; the veins may be dark purple. ( fundus policythaemicus) Facial plethora 1.
POLYCYTHEMIA VERA physical examination Hepatosplenomegaly Erythromelalgia – – – Increased skin temp Burning sensation Redness Liver 40% Spleen 70%
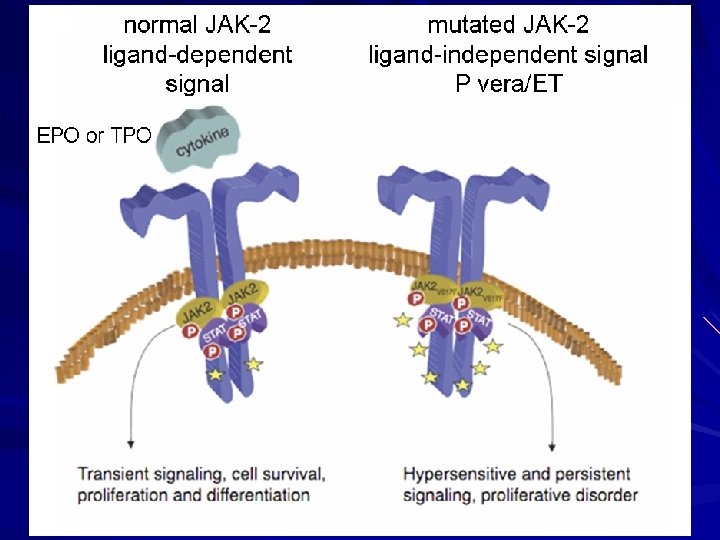
POLYCYTHEMIA VERA Lab Findings CBC – – Hgb/Hct WBC in 45% Plts in 65% Basophilia (seen in all MPDs) Uric acid (can lead to gout) and B 12 Leukocyte alkaline phosphatase score Low epo levels Positive JAK 2 V 617 F
PV - typical blood count WBC x 109/L Hb g/L HCt MCV fl Platelets x 109/L 18. 0 200 0. 62 75 850 Neuts x 109/L Lymphs x 109/L Monos x 109/L 0. 8 Eos x 109/L Basos x 109/L 14. 6 2. 0 0. 1 0. 5 [4 -11] [140 -180] [. 42 -. 51] [80 -100] [150 -450] [2 -7. 5] [1. 5 -4] [0. 2 -0. 8] [0 -0. 7] [0 -0. 1] Film: microcytosis: large and abnormal platelets present
PRV - DIAGNOSIS exclude secondary polycythemia look for features of primary polycythemia measure erythropoietin JAK-2 mutation analysis
SECONDARY POLYCYTHEMIA Arterial blood gas Hb electrophoresis Oxygen dissociation curve EPO level Ultrasound abdomen Chest X ray Total red cell volume(51 Cr) Total plasma volume(125 I-albumin)
Diagnostic Criteria for Primary PV Polycythemia Vera Study Group (PVSG) Criteria for PV Minor Criteria Major Criteria ▪ Elevated RBC mass >36 cc/kg in men >32 cc/kg in women ▪ Oxygen saturation >92% ▪ Splenomegaly ▪ Plt count > 400, 000 ▪ WBC > 12, 000 ▪ Elevated LAP score (>100) ▪ Serum vitamin B 12 >900 pg/m. L or serum unbound B 12 binding capacity >2, 200 pg/m. L → All 3 major criteria OR the first 2 major and any 2 minor criteria ← 2008 WHO Diagnostic Criteria for Primary Polycythemia Vera Major Criteria Minor Criteria 1) Hgb > 18. 5 g/dl (♂) or 16. 5 g/dl (♀) or Hgb or Hct > 99% or Hgb > 17 g/dl (♂) or 15 g/dl (♀) and a documented increase of 2 g/dl or RBC mass > 25% of mean normal 1) Bone marrow trilineage expansion 2) Subnormal EPO level 3) Endogenous erytyhroid colony growth 2) Presence of a JAK 2 V 617 F or similar mutation → two major or first major and two minor criteria ← Tefferi et al. Leukemia (2008) 22, 14– 22
PV - TREATMENT Phlebotomy Myelosuppressive agents – Hydroxyurea – Alkylating agents such as busulfan – 32 P Interferon alpha
PV - PHLEBOTOMY Generally, the best initial treatment for P vera – No increase in progression to AML – Rapid onset – No BM suppression Remove 500 cc blood 1 -2 x/wk to target Hct 45%, then maintain Downsides: – Increased risk of thrombosis – No effect on progression to spent phase – May be insufficient to control disease
PV - MYELOSUPPRETION Hydroxyurea – can be used in conjunction with phlebotomy – May increase the risk of leukemic transformation from 12% to 4 -5% 32 P – increase the risk of leukemic transformation from 1 -2% to 11% – May be appropriate for pts intolerant of medications or for elderly patients – Single injection may control hemoglobin and platelet count for a year or more.
PV – INTERFERON ALPHA Benefits – No myelosuppression – No increase in progression to AML – No increase in thrombosis risk – OK in pregnancy Drawbacks – Must be given by injection – Side effects may be intolerable in many pts, include flu-like symptoms, fatigue, fever, myalgias, malaise
POLYCYTHEMIA VERA I. Patients under the age of 50 with no history of thrombosis and without severe thrombocytosis (greater than 1000 G/L)-phlebotomy alone - initially 450 -500 ml phlebotomy every other day until the hematocrit is less than 46% - older patients or these with underlying cardiovascular disase should undergo smaller phlebotomies 200 -300 m. L twice weekly or 100 -150 m. Levery day until Ht<46% - subsequently, Ht should be mainted between 42 -46% - fluid replacement so that the patients remains isovolemic II. Patients over the age of 70, or with history of thrombosis and with severe thrombocytosis (greater than 1000 G/L)-myelosuppresive agent - Hydroxyurea 15 -30 mg/kg III. Patients 50 -70 years with no history of thrombosis and without severe thrombocytosis (greater than 1000 G/L) individualize therapy I or II 83
POLYCYTHEMIA VERA IV. Antiplatelets agents • Aspirin initially 150 -300 mg/d, maintence therapy 75 -100 mg • Tiklid 2 x 1 • Dipyridamol • Anagrelide 2 -2, 5 mg/d V. Other modalities 1. Radioactive phosphorus (in older than 75 years) 2. Interferon alpha 3 million units 3 times weekly VI. Special Topics 1. Pruritus: antihistaminic agent, cyproheptadine-4 mg three times per day 2. Hyperuricemia-allopurinol 300 mg/day 84
ESSENTIAL THROMBOCYTHEMIA (ET)
THROMBOCYTOSIS Definition: thrombocytosis is defined as a platelet count > 450, 000 cells/μL Etiology of Thrombocytosis Primary - if the thrombocytosis is caused by a myeloproliferative neoplasm, the platelets are frequently abnormal and the patient may be prone to both bleeding and clotting events. Secondary - if thrombocytosis is secondary to another disorder (reactive), even patients with extremely high platelet counts (e. g. , > 1, 000 cells/μl) are usually asymptomatic. Differential Diagnosis of secondary thrombocytosis: 1. Malignancies 2. Infections and inflammatory disorders (e. g. , Crohn’s disease) 3. Post surgical status 4. Connective tissue disorders 5. Iron deficiency anemia 6. Splenectomy 7. Recovery of the bone marrow from a stress (chemotherapy or alcohol) 8. Essential Thrombocythemia
ESSENTIAL THROMBOCYTHEMIA (ET) ET is a clonal myeloproliferative disorder characterized by bone marrow hyperplasia with excessive proliferation of megakaryocytes and sustained elevation of the platelet count. 87
ESSENTIAL THROMBOCYTHEMIA (ET) Neoplastic stem cell disorder causing dysregulated production of large numbers of abnormal platelets Some cases non-clonal (esp young women) Abnormal platelets aggregate in vivo, causing thrombosis Abnormal platelets also cause bleeding
ESSENTIAL THROMBOCYTHEMIA clinical picture 1. Thrombotic complications (intermittent or permanent occlusion of small blood vessels) • transient cerebral and ocular ischemic episodes that may progress to infarction • peripheral arterial occlusive disease associated with „erythromelalgia” (intermittent, painful errythema and cyanosis of the fingers and toes 2. Hemorrhagic complications - bleeding after surgery and spontaneus upper gastrointestinal bleeding (the hemorrhagic tendency is worsened if nonsteroidal anti-inflammatory agent are administered 3. Splenomegaly - 20 -50% patients 4. Hepatomegaly - rarely 89
ESSENTIAL THROMBOCYTHEMIA laboratory findings Thrombocytosis (in most patients>1000 G/l) Numerous thrombocyte aggregates in peripheral blood smear Leukocytosis, usually less than 20 G/l Neutrophilia and a mild shift to the left(usually to metamyelocyte) Slight eosinophilia and basophilia Marked hyperplasia of the megakaryocytes in the bone marrow 90
ET-Typical Blood Count WBC x 109/L Hb g/L MCV fl Platelets x 109/L 10. 0 156 85 1560 [4 -11] [140 -180] [80 -100] [150 -450] Neuts x 109/L Lymphs x 109/L Monos x 109/L Eos x 109/L Basos x 109/L 7. 0 2. 0 0. 8 0. 1 [2 -7. 5] [1. 5 -4] [0. 2 -0. 8] [0 -0. 7] [0 -0. 1] Film Comment: many large and abnormal platelets present
ET – DIAGNOSTIC CRITERIA 2008 WHO Diagnostic Criteria for Essential Thrombocytosis 1. Platelet count > 450, 000 2. Megakaryocytic proliferation with large, mature morphology and with little granulocytic or erythroid expansion 3. Not meeting WHO criteria for CML, PV, PMF, MDS or other myeloid neoplasm 4. Demonstration of the JAK 2 V 617 F or other clonal marker or lack of evidence of a secondary (reactive thrombocytosis) → Diagnosis of essential thrombocythemia requires meeting all four major criteria ← Teferri et al. Leukemia (2008) 22, 14– 22
ET - DIAGNOSIS Criteria of exclusion – – – No evidence of Polycythaemia vera No evidence of CML No evidence of myelofibrosis (CIMF) No evidence of myelodysplastic syndrome No evidence of reactive thrombocytosis Bleeding Trauma Post operation Chronic iron def Malignancy Chronic infection Connective tissue disorders Post splenectomy
ET - OUTCOMES Most patients with ET enjoy a normal life expectancy Like PV, the major risks are secondary to thrombosis and disease transformation: ▪ 15 -year cumulative risks: ▪ thrombosis - 17% risk ▪ clonal evolution into either myelofibrosis (4%) or AML (2%) High risk for thrombosis: ▪ age ≥ 60 ▪ prior thrombosis ▪ long-term exposure to a plt count of > 1, 000
ESSENTIAL THROMBOCYTHEMIA -THERAPY 1. No treatment- asymptomatic( without thrombotic and bleeding complications), young (< 60 r. ż. ) patients with platelet count<1000 G/L 2. Cytoreductive therapy – patients with platelet count>1000 G/L, especially for these with previous thrombotic or bleeding problems - hydroxyurea at doses 15 -30 mg/kg, , to maintein platelet count between 400 -600 G/l 3. Anti-aggregating therapy: Aspirin 75 -150 mg/d dipyridamol for older patients and/or with a cardiovascular risk 4. Anagrelide (Agrylin)- drug that produces selective platelet cytoreduction, and it also inhibits platelet activation doses from 0, 5 mg every 6 hours, to max. 10 mg/d ) 5. Interferon- : 3 million units/d s. c. 97
ESSENTIAL THROMBOCYTHEMIA -THERAPY Low Risk: ▪ Age <60 years ▪ No previous history of thrombosis ▪ Platelet count <1 million/μl → aspirin (81 mg daily) if vasomotor Sx or other medical need for ASA → if otherwise low risk and plt >1. 5 X 106, screen for an acquired von Willebrand disease before instituting ASA High Risk: ▪ Age ≥ 60 years ▪ A previous history of thrombosis → hydroxyurea + aspirin (81 mg daily) → if plt >1. 5 X 106, screen for an acquired von Willebrand disease before instituting ASA → anagrelide is an option, but when c/w hydroxyurea, it was assn with an increased risk of arterial thrombosis, venous thrombosis, serious hemorrhage, or death from vascular causes
MYELOFIBROSIS
MYELOFIBROSIS AKA: agnogenic myeloid metaplasia with myelofibrosis Clonal stem cell disorder affecting megakaryocytes predominantly All myeloproliferative disorders can result in a spent phase which can be difficult to distinguish from primary MF Myeloid metaplasia refers to earlier proliferative phase where extramedullary hematopoiesis predominates.
MYELOFIBROSIS Myelofibrosis is a chronic myeloproliferative disease with clonal hematopoesis and secondary(non-clonal) hyperproliferation of fibroblasts (stimulated by PDGF, EGF, TGF- released from myeloid cells, mainly from neoplastic megakaryocytes) with increased collagen synthesis. It produces bone marrow fibrosis and to extramedullary hematopoesis in the spleen or in multiple organs Other terms – – – agnogenic myeloid metaplasia primary myelofibrosis, osteomyelofibrosis, idiopathic myelofibrosis, myelofibrosis with myeloid metaplasia
MYELOFIBROSIS Insidious onset in older people – asymptomatic (15% - 30%) – severe fatigue Splenomegaly- massive Hepatomegaly Hypermetabolic symptoms – Loss of weight, fever and night sweats Bleeding problems Bone pain Gout Can transform to acute leukaemia in 10 -20% of cases
MYELOFIBROSIS Anaemia High WBC at presentation Later leucopenia and thrombocytopenia Leucoerythroblastic blood film Tear drops red cells Bone marrow aspiration- Failed due to fibrosis Trephine biopsy- fibrotic hypercellular marrow Increase in NAP score JAK 2+ (V 617 F) in approximately 50% of cases
Pathological Features of Peripheral Blood and Bone Marrow in Patients with Myelofibrosis with Myeloid Metaplasia Tefferi A. N Engl J Med 2000; 342: 1255 -1265
MF – DIAGNOSTIC CRITERIA 2008 WHO Diagnostic Criteria for Primary Myelofibrosis Major: 1. Megakaryocytic proliferation and atypia with either reticulin or collagen fibrosis or If no fibrosis, mekakaryocytic expansion must be assn. w/increased BM cellularity 2. Does not meet WHO criteria for CML, PV, MDS, or other myeloid neoplasm 3. Demonstration of the JAK 2 V 617 F mutation or other clonal marker or no other evidence of a reactive marrow fibrosis Minor: 1. Leukoerythroblastosis (immature RBCs and WBCs in the PB) 2. Increased LDH 3. Anemia 4. splenomegaly → Diagnosis of primary myelofibrosis (PMF) requires meeting all three major criteria and two minor criteria ← Teferri et al. Leukemia (2008) 22, 14– 22
MF - OUTCOME q As fibrosis progresses, cytopenias worsen leading to a transfusion dependency ▪ symptoms related to extrmedullary hematopoiesis increase (worsening splenomegaly and ‘B’ symptoms) also are frequently identified q Rarely do patients transform to Acute Leukemia (~ 4%) ▪ clonal evolution was common in these patients ▪ some evidence that in all MPNs, cases of JAK 2 (-) Acute Leukemia arise out of a JAK+ MPN, causing speculation that there additional genetic changes that either initiate and/or propagate these diseases q Despite the lack of transformation to leukemia, three-year survival rate is approximately 52%
MF – RISK ASSASSEMENT Mayo Scoring System (pts age < 60) Score Median Survival 0 173 mo 1 61 mo ≥ 2 26 mo Transplant Scoring System (pts age < 55) Score Median Survival 0 or 1 15 yrs ≥ 2 3 yrs Risk Factors: Hemoglobin <10 g/d. L White blood cell count <4000/μl or >30, 000/ μl Absolute monocyte count >1000 μL Platelet count <100, 000/ μL Risk factors: Hemoglobin <10 g/d. L ‘B’ symptoms present (eg, fever, NS, weight loss) Circulating blasts >1 percent Elliott et al. Leuk Res. 2007; 31(11): 1503 -9. Dupriez et al. Blood 1996 Aug 1; 88(3): 1013 -8.
MYELOFIBROSIS - TREATMENT Androgens(oxymetholone 2 -4 mg/kg) in anemia from decreased red cell production -overall response is about 40% Cortykosteroids(prednisone 1 mg/kg) in anemia with shortened red cell life-span-response in 25 -50% of patients Hydroksyurea (15 - 20 mg/kg) for the control of leukocytosis, thrombocytosis, or organomegaly Allopurinol-to prevent hyperuricaemia Vit. D 3 -analogues(1, 25 -dihydroxycholecalciferol-1 ug/d (? ) Transfusions of packed red cells for anemia or platelets for thrombocytopenia with bleeding 108
MYELOFIBROSIS - TREATMENT Splenectomy should be considered for: portal hypertension, painful splenomegaly, refractory anemia and thrombocytopenia, or exccessive transfusion requirement. However, the procedere is hazardous (an operative mortality is up to 38%). Splenic irradiation: when there is a contrindication to splenectomy Allogeneic stem-cell transplantation: for young patients who have a poor prognosis and have a suitable donor identified. Experimental therapies: Interferon- , antifibrotic and antiangiogenic drugs (anagrelide, suramin, pirfenidone, thalidomide, ) 109
MF - TREATMENT Risk stratification is critical in deciding on therapeutic options (see previous scoring systems) ‘Low Risk’ without symptoms – expectant management ‘Low Risk’ with symptoms – hydroxyurea androgenic and corticosteroids splenectomy if adequate BM hematopoiesis splenic irradiation thalidomide or lenalidomide ‘High Risk’ and age < 55(? ) – consider a reduced intensity allogeneic BMT
MYELOFIBROSIS - prognosis - a median survival of 3, 5 to 5, 5 years - the principal causes of death are infections, thrombohemorrhagic events, heart failure, and leukemic transformation - leukemic transformation occurs in approximately 20% of patients during first 10 years 111