Unit 2 Organic Chemistry and Instrumental Analysis Revision



























.")




")






















, amides, nitriles and")



























































- Slides: 116
Unit 2 Organic Chemistry and Instrumental Analysis
Revision Ø Be able to define the following: Homologous series Functional group Saturated Unsaturated Isomers Addition reaction Condensation reaction Hydrolysis Physical properties Chemical properties
Covalent Bonding Ø A covalent bond forms when atomic orbitals overlap to produce a molecular orbital. Ø This new arrangement of electrons and nuclei is of lower energy than before bonding occurred. Attractions and repulsions are equal. Ø A chemical bond can be thought of as the localisation of electrons holding together 2 adjacent positive nuclei.
Covalent bonding continued
Ø On the graph above the energy required to separate the atoms is the bond enthalpy and r 0 is the length of the bond. Ø Chemical bonds are a continuum from covalent to ionic. Electronegativity values can help to predict the type of bonding, but polar covalent bonding has many forms. This does not account for metallic bonding or exceptions like graphite.
Bonding in alkanes Ø When carbon has 4 single bonds it has a tetrahedral arrangement. Ø The bonds are covalent. Carbon has electronic structure 1 s 2 2 p 2 or to use box notation: 1 s Ø 2 s 2 p 1 s sp 3 hybrid The 2 s and 2 p orbitals combine to form “sp 3” hybrid orbitals.
Hybrid orbitals Ø sp 3 hybrids are made of 1 x 2 s and 3 x 2 p orbitals. Ø Electron repulsion theory agrees with the tetrahedral arrangement. Ø The shape is a distorted combination of an s and a p orbital.
Sigma bonds Ø Overlap of a half filled sp 3 with a half filled s orbital creates a sigma bond. Ø The overlap is end on and along the axis joining the 2 nuclei. Ø A sigma bond can also be formed from the overlap from overlap of 2 hybrid orbitals to form a C-C single bond. Ø All alkanes have bond angles of 109. 5° – tetrahedral angle.
Bonding in alkenes Ø Ethene is flat with angles of 120°. The C=C is shorter than C-C, but not double the strength. The C=C cannot rotate easily. Ø This time the 2 s and 2 x 2 p orbitals form 3 hybrid orbitals ie “sp 2”. Ø The sp 2 orbitals lie at 120° to each other and at 90° to the remaining p orbital. Overlap creates 1 x sigma and 1 x π bonds (overlap of 2 parallel sp 2 hybrids).
Bonding in alkynes Ø Alkynes contain a Carbon to Carbon triple bond. Ø This l is formed from SP hybridisation i. e. 1 s and 1 p orbital overlap end on, leaving the 2 remaining p orbitals to overlap side on in different planes to create the triple bond.
Stereoisomers Ø Often isomerism can be subtle. Ø Stereoisomers have the same molecular and structural formulae, but the atoms have different 3 D arrangements, making them nonsuperimposable on each other. Ø Your hands are an example. They have the same number of digits, the same connections, but no matter how you try and turn them, you cannot make them identical.
Geometric isomers Ø These isomers occur when there is no free rotation around a C-C bond ie this often a C=C bond. Ø Two kinds – cis and trans. Ø The bond is not easily broken to allow rotation.
Physical properties of geometric isomers Ø Often have different melting points – trans higher than cis. Ø This is due to trans isomers being able to pack more closely, therefore they have stronger intermolecular forces of attraction. Ø Try Q 4 -13 pg 127 -129 Scholar book 3.
Chemical properties of geometric isomers Ø Sometimes geometric isomers can have different chemical properties. Ø Eg butenedioic acid. The cis form arrangement allows for water to be lost under heating to form a cyclic anhydride. Ø The arrangement of the trans form does not allow this.
Note the significant different in melting points in the cis and trans forms. Can you draw the structure of the product?
Optical isomerism Ø Occurs when there are 4 different groups around a tetrahedral carbon atom. Ø Called chirality – arises from a lack of symmetry. Ø Eg a right hand glove does not fit a left hand vice versa. Ø The isomers cannot be lined up or superimposed on each other.
Enantiomers Ø Each optical isomer is called an enantiomer. Ø When the enantiomers are 50% each the mixture is called a racemic mixture or racemate. Ø This was made famous with thalidomide – the racemate was prescribed instead of a single enantiomer. Ø Many biological systems use only one enantiomer eg alanine and other amino acids.
Alanine enantiomers Ø Use a modelling kit to build the enantiomers of alanine and check that they cannot be superimposed.
Polarised light Ø Enantiomers rotate plane polarised light to the same degree in different directions. This is why they are called optically active. Racemates are optically inactive i. e. the rotation from each enantiomer cancels out, giving no overall rotation. Ø Although they behave the same in test tube reactions, they behave differently in the presence of other chiral molecules. Ø Eg 1 enantiomer of limonene smells of lemons, the other of oranges.
Nucleophilic substitutions Ø Chiral compounds produce chiral products. Ø SN 2 reactions give rise to an inverted product – see later notes on SN 2 reactions. Ø SN 1 reactions give rise to racemic mixtures because the nucleophile can attack from either side. Ø Try Q 22 -23 pg 142 and Q 25 -26 pg 144 Scholar book 3.
Bond breaking Ø Homolytic fission – 2 shared e separate equally. Highly - reactive free radicals form. Ø Heterolytic fission – both shared e- go to 1 atom. Ions are produced. Ø Note – pi bonds eg in alkenes and carbonyls are easier to break than sigma bonds due to the side on overlap rather than end on. This makes them important in synthesis.
Carbocations Ø These species form during heterolytic bond fission eg Ø The bonds undergoing heterolytic fission tend to be polar. Ø The electrons go to the more electronegative species
Nucleophiles Ø “nucleus seeker” Ø Negatively charged ions eg OH-, carbanions attracted to positively charged nuclei. Ø Atoms with partial negative charge can also be nucleophiles eg nitrogen in ammonia.
Electrophiles Ø “electron seeker” Ø Positively charged eg H+, carbocations attracted to negatively charged electrons. Ø Partially positive atoms can act as electrophiles eg hydrogen atoms in water. Ø Polar molecules can therefore have both an electrophilic and a nucleophilic centre.
Halogenoalkanes Ø Can be called alkyl halides or haloalkanes. Ø Fluorine gives prefix “fluoro” etc. Ø Ø Name longest chain. Give substituents lowest possible number. List halogens in alphabetical order. Then list other substituents eg alkyl. Ø Try Q 21 and 22 pg 32 Scholar book 3.
Types of halogenoalkanes Primary Secondary Tertiary Try Q 23 -26 pg 33 Scholar book 3
Elimination reactions Ø Strong base eg KOH in ethanol. Ø OH- attacks hydrogen atom of halogenoalkane. The H-C bond breaks, creating a new C=C bond and the halide atom leaves as a halide ion. Ø Try Q 27 -30 pg 35 Scholar book 3.
SN 1 reactions Ø Substitution, nucleophilic, 1 st order (1 molecule involved in RDS). Ø SN 1 reactions generally only occur with tertiary halogenoalkanes, due to the stability of the
Mechanism of SN 1
The inductive effect Ø To stabilise the carbocation, alkyl groups push electron density towards the positively charged carbon atom. Ø This stabilises the positive charge. Ø Therefore SN 1 tends only to happen with tertiary halogenoalkanes.
SN 2 reactions Ø Substitution, nucleophilic, 2 nd order (rate depends on the concentration of both reactants). Ø 1 step reaction, no intermediate, transition state forms. Reaction can go backwards or forwards from the transition state. TS cannot be isolated.
Mechanism for SN 2
Products of substitution reactions Ø Add OH- to make alcohols. Ø Add alkoxides (RO-) in sodium and ethanol to make ethers (R-O-R). Ø Add – to make nitriles and extend the carbon chain by 1. Ø Add ammonia to make amines. Ø Try Q 36 -39 pg 41 Scholar book 3.
Alcohols and ethers Ø Alcohols – functional group = OH Ø Used to make plastics, solvents, drinks, antifreeze, steroids, fuels, explosives. Ø Ethers – functional group = R-O-R Ø Used as anaesthetics, solvents and petrol additives. Ø Try Q 5 -8 pg 47 Scholar book 3.
More on ethers Ø Can be written as R’-O-R’’ or R 1 -O-R 2. Ø If R’ and R’’ are identical – symmetrical ether. Named as “dialkylether”. Ø If R’ and R’’ different – unsymmetrical ether. Named as substituents eg methoxyethane. Smaller alkyl group is the substituent. Ø Try Q 9 -12 pg 48 Scholar book 3.
Preparing ethers Ø Reaction of metal alkoxide with monohalogenoalkane.
Reactions of ethers Ø Slightly polar, but not enough to be attacked by nucleophiles and electrophiles. Ø Highly flammable!! Unstable and explosive. Ø Form hydroperoxides and peroxides on contact with air over a few days. Ø Good solvents for polar and non-polar substances. Eg ethoxyethane easily evaporated, but exercise caution. Ø Treat ethers with an iron salt to ensure peroxide-free solvent.
Physical properties Ø Complete the word bank on pg 49 Scholar book 3. Then try Q 13. Ø Consider the forces of attraction between alkanes, alcohols, ethers and halogenoalkanes. Use your knowledge to answer Q 14 -26 pg 50 -52 Scholar book 3. Ø Due to lack of hydrogen bonding, ethers have lower bpts than alcohols. Ø Now try the solubility questions pg 52 -53 Scholar book 3.
Preparation of alcohols Ø Traditionally by fermentation. Ø By hydration of alkenes. Good for large scale production. Ø By preparation from halogenoalkanes – SN 1 or SN 2 with OH- nucleophile. Generally only 1 product formed.
Reactions of alcohols Ø Alcohols react with alkali metals to create metal alkoxides and hydrogen gas (similar to reaction of water with alkali metals to make hydroxides and hydrogen gas). CH 3 OH + Na CH 3 O-Na+ + H 2 Ø Metal alkoxides, RO-, are powerful bases and nucleophiles.
Dehydration of alcohols Ø Creates alkenes. Al 2 O 3 catalyst Ø More generally alcohol + concentrated H 2 SO 4 or H 3 PO 4 gives alkenes.
Synthesis of alkenes – elimination reactions Ø Dehydration of an alcohol. Alcohol vapour passed over Al 2 O 3 catalyst to produce water and an alkene. Ø Alternatively for less volatile alcohols the alcohol is warmed to 80°C with phosphoric or sulphuric acid. Ø Another method is to remove hydrogen halide from a monohalogenoalkane using KOH in ethanol. Ø Most reactions produce a mixture of products.
Reactions of alkenes – electrophilic addition Ø The C=C bond in alkenes is electron rich ie nucleophilic. It can donate a pair of electrons to an electrophile. l l Catalytic addition of hydrogen Halogens to form dihaloalkanes Addition of hydrogen halides (HCl) Acid catalysed addition of water
Electrophilic addition 1
Electrophilic addition 2
Electrophilic addition of hydrogen halides Ø These molecules are polar. The δ+ hydrogen atom is attacked by the π electrons and the H-Cl bond breaks heterolytically. Ø The Cl- ion then attacks the carbocation to create a monohalogenoalkane. Ø For ethene only one product can be formed as the molecule is symmetrical. However longer chain alkenes can form different products.
Markovnikov’s rule Ø Attack of propene could result in either 1 chloropropane (attack at the end of the chain) or 2 -chloropropane (attack at the middle carbon atom). Ø Mostly the product is the latter. Ø Markovnikov’s rule states that H adds to the carbon atom with the least alkyl groups and the halogen adds to the carbon atom with more alkyl groups.
Stability of carbocations Ø Markovnikov’s rule can be explained by looking at the stability of the carbocations formed. Ø The stability is conferred by alkyl groups pushing electron density towards the positive carbon centre and therefore stabilising the positive charge.
Acid catalysed addition of water Ø First the double bond attacks the H+ ion. Ø Then the δ– oxygen atom of water attacks the carbocation. Ø X- from the acid then removes a proton from the water molecule, giving oxygen back the lone pair of electrons and forming HX. Ø See preparation of alcohols for mechanism.
Addition with halogens Ø Unlike hydrogen halides and water, diatomic halogen molecules are non-polar. Ø As the halogen molecule approaches the electron rich double bond of the alkene, the electrons of the halogen bond are repelled, creating a temporary dipole. Ø The addition then proceeds via an unstable cyclic intermediate, followed by nucleophilic attack of the generated halide ion.
Addition with halogens
Catalytic addition of hydrogen Ø Used in fat hydrogenation. Ø Catalyst is usually transition metal eg Pt, Pd, Ni. Ø Hydrogen adsorbed onto catalyst surface and broken into atoms. Ø The alkene π bonds to the catalyst surface. Then nearby hydrogens can bond to the alkene, which loses its π electrons and leaves the surface.
Carbonyl group Ø Aldehydes, ketones and carboxylic acids. δ- Ø C=O similar to C=C. Ø Oxygen orbitals hybridise, however oxygen is more electronegative than carbon so the C=O bond is polarised. Ø This makes an electrophilic carbon centre and a nucleophilic oxygen centre. δ+
Properties of carbonyl compounds Ø Boiling points can be considered in the same way as alcohols and ethers. This time it is the polar-polar interactions that affect the boiling points. Ø Try Q 7 -10 pg 69 Scholar book 3. Ø Solubility in water of organic compounds depends on hydrogen bonding ability. Ø Try Q 11 -14 PG 71 Scholar book 3.
Acidity Ø In aqueous solution carboxylic acids partially dissociate. p. Ka values can be used to measure the strength of the acid. Ø The alkanoate ion can delocalise: Ø Unhybridised p orbitals overlap to make a delocalised π system. Ø Try Q 17 -19 pg 73 Scholar book 3.
Carboxylic acids Ø Can be made from 1° alcohols (via aldehydes), amides, nitriles and esters. Ø Can react with metals and metal compounds to form salts eg sodium ethanoate. Ø React with bases like ammonia and amines to make salts eg ammonium propanoate. These salts can be heated to produce amides. Ø Try Q 38 -40 pg 84 Scholar book 3.
Oxidation of aldehydes Ø Aldehydes oxidise to carboxylic acids. This is because they have a hydrogen atom on the carbonyl carbon and ketones do not. Ø Tollens’ reagent – [Ag(NH 3)2]+. Silver mirror test as silver ions reduced to silver metal. Ø Fehling’s solution – copper (II) ions reduced to copper (I) ions and a red/brown precipitate of copper (I) oxide forms.
Reduction of carbonyls Ø Aldehydes give 1° alcohols Ø Ketones give 2° alcohols Ø Uses Li. Al. H 4 in ether. Creates a hydride ion to attack the carbonyl carbon.
Nucleophilic addition Ø First the carbonyl carbon is attacked by a nucleophile, then the O- is protonated. This is similar to the reduction mechanism. Hydrogen cyanide cyanohydrins Ø Hydrazine NH 2 -NH 2 hydrazones Ø 2, 4 -dinitrophenylhydrazine (Brady’s reagent) 2, 4 dinitrophenylhydrazones Ø
Ester formation Ø Alcohol adds nucleophilically to the carbonyl of the carboxylic acid. Ø The oxygen of the carbonyl is protonated. Ø Water is then lost across the carbonyl group to form the ester. Ø The reaction is reversible.
Esterification mechanism
Reduction of carboxylic acids Ø Use Li. Al. H 4. Ø More resistant to reduction than aldehydes and ketones. Ø Nucleophilic attack by H- ion. Ø Try Q 46 -52 pg 87 Scholar book 3.
Amines Ø Smell of fish – found in decaying matter. Ø Functional group of amino acid. Ø Polyamides form eg nylon, kevlar. Ø Polypeptides – many amino acids. Then form proteins. Ø Similar properties to ammonia.
Naming amines Ø If amine is the only functional group, then amine is added to the substituent group eg propylamine. This can also be called propanamine. Ø If the amine is a side chain the prefix amino is used eg 2 -aminobutane. Ø Prefixes and numbering is as with other compounds. Ø Try Q 1 -2 pgs 91 -92 Scholar book 3.
Classification Ø 1° – 2 H atoms on the N of the amine. Ø 2° – 1 H atom Ø 3° – No H atoms Simplest aryl amine is aniline or phenylamine (has benzene as the R group). Ø Ø Try Q 3 -6 pgs 94 -95 Scholar book 3.
Physical properties Ø Properties influenced by polar N-H bonds. Ø 1° and 2° have hydrogen bonding, but 3° only have van der Waals. Ø Amines therefore have higher boiling points than alkanes of similar molecular mass. Ø Water can hydrogen bond to all amines, therefore those with small R groups can be soluble in water. Ø Try Q 7 -13 pg 96 Scholar book 3.
Chemical properties Ø Strongly influenced by lone pair on nitrogen. Ø Proton acceptors ie bases. Ø Dissolve in water to make alkalis – an ammonium ion and hydroxide ion produced. Ø Copy fig 5. 8 pg 98 Scholar book 3. Ø Weak bases. Use p. Ka of conjugate acid to determine basicity. Higher the p. Ka, the stronger the base. Ø Try Q 14 -16 pg 98 Scholar book 3.
Salt formation Ø Amines react with acids to produce salts. Ø This is a neutralisation reaction. Ø They react with carboxylic acids to make salts, which can then be heated to make amides – see previous chapter. Ø Try Q 17 -18 pg 99 Scholar book 3. Ø Then try Q 19 -25 pg 102 Scholar book 3.
Aromatics Ø Traditionally sweet odours. Ø Benzene = C 6 H 6 Ø Phenyl group = C 6 H 5 - Ø Alkyl-substituted aromatic hydrocarbons are called arenes. Ø Unsaturated, but do not do addition reactions like alkenes. Instead they undergo electrophilic substitutions.
Benzene structure Ø Friedrich Kekulé proposed an alternating single and double bond system where the bonds oscillated between the 2 possible structures. Ø This gives rise to the more common representation:
More on benzene structure Ø sp 2 hybridised carbons in a flat symmetrical structure. There are sigma bonds from each carbon to its hydrogen. Ø Each C atom has a p orbital perpendicular to the plane of the ring with 1 π e-. These overlap to produce a delocalised π e- system. Ø This creates a doughnutshaped area of negative charge above and below the plane of the ring. Ø Try Q 1 -6 pgs 105 -106 Scholar book 3.
Electrophilic substitution Ø The high electron density of benzene attracts electrophiles. Ø The electrophile attacks, then the hydrogen atom is lost to restore the π system. Ø The intermediate positive charge is stabilised by the delocalisation.
Halogenation Ø Catalyst – Fe. Cl 3, Fe. Br 3 or Al. Cl 3 polarises the diatomic halogen molecule. Ø The partially positive halogen atom attacks the benzene to form the positively charged intermediate. Ø Then the C-H bond breaks heterolytically and the hydrogen atom forms a hydrogen halide to regenerate the catalyst.
Nitration Ø Nitrating mixture – concentrated nitric and sulphuric acid which generates the nitronium ion NO 2+. Ø The nitronium ion attacks the benzene ring and creates an intermediate similar to the halogenation reaction. Ø This creates nitrobenzene, important for dyestuffs. Ø Above 55ºC di and tri-nitrobenzenes can be formed.
Sulphonation Ø Reacts with conc H 2 SO 4 under several hours reflux. Ø Or with H 2 SO 4 and SO 3 under cold conditions. Ø +SO H 3 is reactive species (electrophile).
Alkylation Ø Sometimes called Freidel-Crafts reaction. Ø Al. Cl 3 polarises a halogenoalkane so the alkyl chain can be added to the benzene ring. Ø Try Q 20 -24 pgs 116 -117 Scholar book 3.
Elemental Analysis Ø Reminder - % C, H, N and S in a compound can be found by burning a known weight of a compound in oxygen. Ø This can then be used to determine the empirical formula of the compound. Ø Divide each percentage by the mass of that element to give a mole ratio, then simplify to obtain whole numbers. Ø See example on pg 149 Scholar book 3. Then try Q 2 -8 pg 149 -150 Scholar book 3.
Mass Spectrometry Ø Needs only a small amount of sample. Ø The sample is vapourised, bombarded with electrons to produce positive ions, focused and then passed through a magnetic field to a detector. The electron beam must have greater energy than the molecular ionisation energy. Ø A spectrum of abundance of the various ions is produced as a mass/charge (m/z) ratio.
Interpreting spectra Ø When molecules are hit with electrons, they can either gain or lose an electron. Ø M + e- M. - or M + e- M. + + 2 e- Ø The radical electrons are often left out and it is normally the positive ions that are analysed. Therefore the molecular ions are called M+.
Ion fragmentation Ø Molecular ions can either combine with other ions to form neutral molecules or other ions. Neutral and negative ions are not detected. Ø The highest peak is usually the molecular ion. The height of the peaks on the mass spectrum indicate abundance levels. Ø The most abundant peak is called the base peak and is assigned abundance 100%. Ø Try Q 9 -10 pg 152 Scholar book 3.
Fragmentation patterns Ø Molecular ions can often be unstable and break down into fragments. These fragments can be used to put together a picture of the molecule, like doing a jigsaw. Ø Some aromatic compounds are very stable and can form M 2+ ions. As the peak measures m/z, a peak can be seen at half the value of the mass of the molecular ion. Ø Other compounds are very unstable and only fragment ions are seen. Ø See examples pg 153 -154 Scholar book 3. Try Q 11 -12 pg 154 Scholar book 3.
Using fragmentation patterns Ø Fragmentation patterns can be used to infer stability of different bonds, by studying how the molecule breaks up. Ø Using a standard electron beam strength means a database of spectra has been created so unknown samples can be compared electronically to identify them. Ø Loss of a methyl group is often written as (M-15)+ to show that a CH 3 group has been lost from the molecular ion. Ø Work through the example on pg 155 -160 and Q 14 -21 pg 158 -160 Scholar book 3.
High Resolution Mass Spec Ø This can differentiate between isotopes. Normal mass spec only uses the average RFM values. Ø This was initially used to measure isotopic masses. Ø Molecules with very similar masses can be distinguished by using accurate masses. Ø Try Q 22 -33 pg 160 -162 Scholar book 3.
Infrared Spectroscopy Ø IR radiation absorbed by skin excites skin molecules and can be used to create thermal images. Ø This can also be used for “night vision” and studying cloud cover. Ø Molecules also absorb IR radiation and by measuring IR frequencies and absorbance functional groups can be identified. Ø IR = 1014 - 1013 Hz which corresponds to 200 - 4000 cm-1.
IR interactions Ø Simple diatomic molecules just stretch and compress as they only have 1 bond. Ø The heavier the atoms the less the vibration. Ø Each molecule has a specific frequency that it resonates at. Ø When radiation of varying frequencies is passed through a sample most will pass through intact. The frequency used by the molecule is absorbed and the molecule vibrates in a higher quantum state. The energy is lost in collisions and the molecule falls back to a lower state.
Further vibrational modes Ø Water can have symmetric and assymetric stretches. Ø This gives rise to 2 different IR absorptions. Ø There also bending and twisting modes each with their own frequencies. Ø Stretches require more energy than bending modes.
The IR spectrometer Ø The IR beam is split in 2, one passes through the sample, the other through the reference. Ø The beams then pass through a monochromator grating, then on to the detector which compares the intensity of the 2 beams. Ø A spectrum of % absorbance or transmission is plotted. Ø The sample is put in a nujol paste and then placed between 2 Na. Cl or KBr discs. These compounds do not affect the absorbance.
Interpreting spectra Ø Clear peaks can often be seen at higher wavenumbers. Ø Lower wavenumbers give rise to the fingerprint region – an area of much smaller peaks. Ø A list of absorbances can be found in the data book. Ø Try Q 8 -12 pg 176 -178 Scholar book 3.
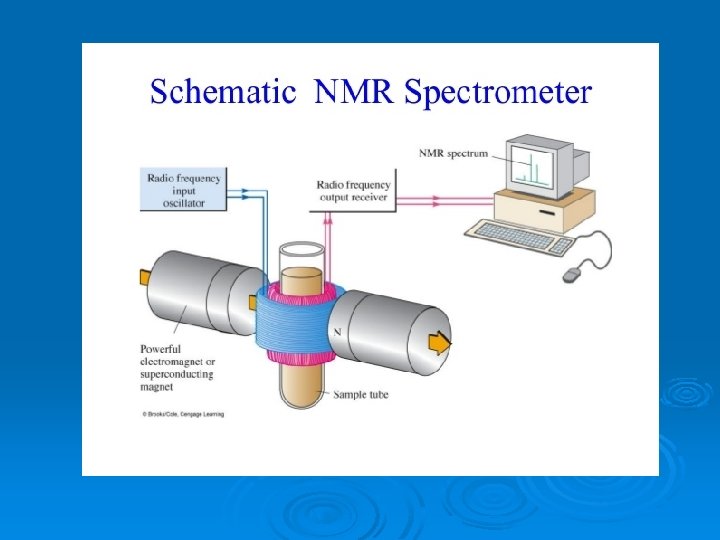
Nuclear Magnetic Resonance Ø Some atomic nuclei have a nuclear spin – like an electron spin. They behave like tiny magnets. Ø In a strong magnetic field the spins align up or down. Ø Applying em radiation makes some nuclei flip to a higher state, absorbing the energy corresponding to the radiation applied. Ø EM radiation is usually radio – 60 -1000 MHz. Ø Common nuclei are 1 H and 13 C.
The NMR spectrometer Ø The sample is mixed with a deuterated solvent. This is to stop the spectrum being swamped with 1 H resonances. Ø The sample is loaded into the machine and suspended in the magnetic field. Ø The radio frequency is applied and then detected to produce a spectrum. Ø The magnet has to be supercooled with liquid helium and takes weeks to set up or shut down.
Spectral analysis Ø The spectra are measured in ppm against a TMS standard. Ø Nuclei in different environments can be detected eg a CH 2 is different to a CH 3. Ø Integration of the area under a peak in a 1 H NMR spectrum gives the number of hydrogen atoms present in that peak. Ø Each peak is shifted relative to TMS. This is called chemical shift, and gives information on the environment of the nuclei. Ø Try Q 20 -29 pg 182 -184 Scholar book 3.
High resolution NMR Ø Often the spectra seen have peaks split into doublets, triplets and quartets. These can overlap giving rise to multiplets and more complex spectra. Ø This happens because the protons on adjacent carbons interact with each other and split the signal coming from their neighbour. Ø The best way to understand NMR is to practise interpreting spectra.
Consider the 1 H NMR spectrum of chloroethane
Ø The signal at ~1. 2 ppm from the methyl CH 3 is split by each of the protons in the CH 2 group, giving a triplet. The second protonsplitsthe both signals Ø The first proton signal into a doublets, doublet. which overlap to create the triplet observed.
Ø The signal at ~ 3. 3 ppm is created by the signal from the CH 2 being split by each of the 3 protons from the CH 3. The CH 2 is shifted due to its proximity to the Cl. Ø Each proton in turn splits the original signal to create a quartet.
Try to interpret this spectrum of ethanol. Explain the patterns and the chemical shifts observed.
Try to interpret this spectrum of the ester ethyl acetate. Explain the patterns and the chemical shifts observed.
MRI Ø Magnetic resonance imaging uses water molecules in the body. Ø Different tissues in the body shield the protons in water to different extents leading to different absorbances of radiofrequency. Ø Data collected can then be used to produce a picture of inside the body without being harmful to the patient.
Paracetamol NMR
Colours of organic compounds Ø Colours in transition metal complexes are due to electrons moving between d orbitals. Ø So why are some organic compounds coloured? Ø Electrons can be thought of as waves. Waves can be in phase or out of phase (constructive and destructive interference). Ø If electrons are in orbitals, then orbitals can be thought of as in phase or out of phase.
Ø We already know that atomic orbitals overlap to make molecular orbitals. If this happens in phase then we create a bonding molecular orbital, where the electrons are found. Ø If it happens out of phase an empty antibonding or unoccupied orbital is created. Ø Electrons can be promoted to the lowest unstable antibonding orbital from the bonding orbital which is closest to it in energy.
As electrons fall back down they emit light of a certain wavelength. Ø This can happen with sigma and pi bonds. Ø
The orbital the electrons are promoted from is called the HOMO (highest occupied molecular orbital). Ø The antibonding orbital they move to is called the LUMO (lowest unoccupied molecular orbital). Ø
Conjugation Ø For small molecules eg methane, the gap between HOMO and LUMO is still quite large, so the absorbtion is in the UV range. Ø For molecules with a series of alternating double and single bonds, there are more LUMO possibilities created and the energy gap is smaller. Ø The more conjugation, the smaller the energy gap. Ø Smaller energy means longer wavelength so these compounds can exhibit visible colours.
Chromophores Ø A chromophore is the part of the molecule responsible for the colour.
Examples of conjugation and chromophores Ø Vitamin A – absorbance ~440 nm. Absorbs dark blue, appears yellow. Ø Indigo – absorbance ~602 nm. Absorbs orange, appears blue. Ø Azo dyes contain N=N and this enables vivid colours eg reds and yellow to be made.
Medicines Ø Pharmacologically active drugs with beneficial properties. Ø Drug – substances which alters the normal biochemical processes in the body. Ø Eg alcohol, caffeine, antibiotics, aspirin.
Drug development Ø Compounds from plant and animal extracts were traditionally used as medicines. Ø These have now been developed so they are produced on a large scale and are more effective and with less side effects. Ø Morphine is extracted from opium poppy juice. Codeine and methadone are derivatives of morphine.
How a medicine functions Ø Medicines interact with the body by binding to receptors made of proteins. These are found on cell surfaces. Ø This can involve London dispersion forces, hydrogen bonding or ionic bonding, but the bonding is usually reversible. Ø The orientation, shape and size of the molecules is important to achieve binding. Ø A biological response eg a muscle stimulus is triggered by the binding.
Ø Effective medicines work by mimicking the natural molecule or blocking the active site. Ø If it mimics the natural molecule it will trigger the same response and is called an agonist. Eg salbutamol which mimics adrenaline and opens the airways. Ø If the drug blocks the active site it prevents the response and is called an antagonist. It is used to block a message eg pain. Eg propranolol blocks adrenalin receptors in the heart to relieve high blood pressure.
Enzyme function Ø Enzymes are large protein molecules which act as biological catalysts. Ø They have 3 D sites called active sites of catalytic receptors. This has a specific shape and will therefore only bind to a specifically shaped substrate. Ø Medicines can block the active sites of enzymes – enzyme inhibitors – to switch the enzyme off.
Other drug targets Ø As well as receptors and enzymes, drugs can target DNA and RNA. Ø They can interfere with the mechanism of replication. Ø This can be useful for anticancer or antiviral medicines. Ø Eg methotrexate which prevents the formation of DNA base precursors.
The pharmacophore Ø Minimum structural feature that gives pharmacological activity. Ø The shape fits the receptor site and other functional groups assist binding to the site. Ø Once this is established, modifications can be made to the pharmacophore to improve its performance as a medicine and reduce side effects. Ø Read through the case studies pg 199 -207 Scholar book 3.
% by mass and volume and ppm Ø To make a % by mass solution simply weigh out the required mass and make up to 100 cm 3 with distilled water. 3 l Eg 1% starch is 1 g starch in 100 cm water Ø % by volume is the number of cm 3 of solute made up to 100 cm 3 of solution. 3 3 l Eg 20% vol H 2 O 2 is 20 cm in 100 cm Ø The unit ppm stands for parts per million and refers to 1 mg per kg.