Chapter 2 The first law of thermodynamics thermodynamics
 energy conversion")



")
 the infinitesimal changes of a state function")





 is the exchange of thermal")

 container filled")
: All the transferring form of energy except heat. • There")

















 •")
 • Does a temperature has an absolute zero, or 0")







= δQ/d. T")















 from a solid to")


















 + 2 H")



+2 O 2(g) CO 2(g)+2 H 2 O(g) c.")
 的系统,含氧气 4 mol ,氮气(4/0. 21)× 0. 79 mol=15. 05")





 Throttling:d H=0)")








 (adiabatic reversible) p. Vγ=const (adiabatic reversible for ideal gas)")


 2. 2 (basic concept)")
 • 2. 25 (Calculation of the")
- Slides: 111
Chapter 2 The first law of thermodynamics • thermodynamics deals with (a) energy conversion and (b) direction of change. • Two laws, the first law and the second law of thermodynamics, are foundation of thermodynamics. • They are an experience summary of human beings in long history, therefore can not be proved mathematically, while their correctness is indubitable.
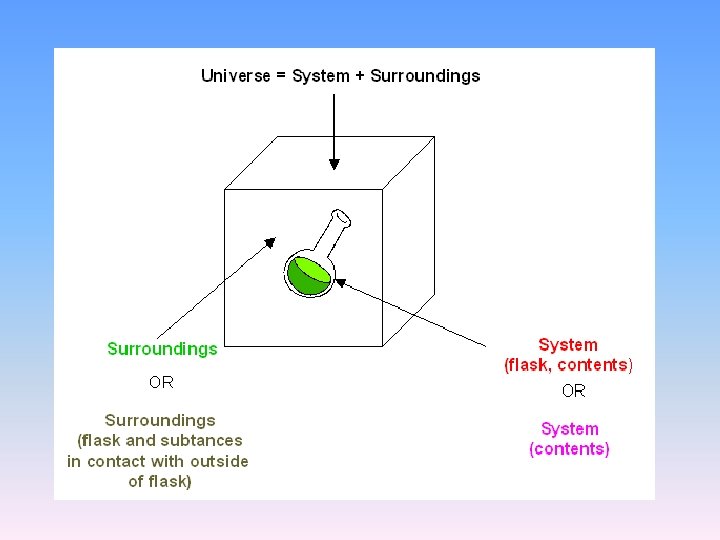
2. 1 Concept and nomenclature in thermodynamics • 2. 1. 1 System and surroundings
Three kinds of systems • Open systems: • exchange of both matter and energy • Closed systems: • no exchange of matter but some exchange of energy. • Isolated systems: • neither exchange of matter nor exchange of energy.
2. 1. 2 Extensive property and intensive property • Extensive property • its value depends on the extent or size of the system. The overall value is the sum of various parts of the system. • For example, m, V, U, etc. • Intensive property • Its value is independent of the extent or size of the system. • For example, T, c, ρ, etc. • any extensive variable divided by the moles or mass becomes an intensive variable.
2. 1. 3 State and state function • State • 状态是指系统的各种内在及外在性质在 一定条件下的宏观表现。(化 2004一同 学) • State function • a property of a system that is not dependent on the way in which the system gets to the state in which it exhibits that property.
Two properties of state functions • (1) the infinitesimal changes of a state function can be expressed in total differential. e. g. z=f(x, y) • (2) Any changes between the initial and final states depend only on the state of the system not on the paths through which the change takes place.
ΔT=T 2-T 1, ΔU=U 2- U 1 ΔY=Y 2-Y 1, ΔX=X 2-X 1 Y A B X
2. 1. 4 Equilibrium state • three conditions have to be necessary to an equilibrium state: • (A) Thermal equilibrium • (B) Mechanical equilibrium • (C) Chemical reaction and phase transition equilibrium
2. 1. 5 Steady state • Steady state is a situation in which all state variables are constant in spite of ongoing processes that strive to change them. • It is different from the equilibrium state.
2. 1. 6 Process and path • Process is a change of a system from initial state to finial state. • Path is the intermediate steps between the initial state and the final state in a change of state. • Isobaric: process done at constant pressure, p 1=p 2=psur. • Isochoric: process done at constant volume, V 1=V 2. • Isothermal: process done at constant temperature, T 1=T 2=Tsur. • Adiabatic: process where Q = 0, that is, no heat exchanges • Cyclic: process where initial state = final state.
Spontaneous and non-spontaneous • A spontaneous process is one that will naturally occur in the absence of external driving forces. For example, a ball rolls off a table and falls to the floor. • A non-spontaneous process is the reverse of a spontaneous process. This does not mean that nonspontaneous processes do not happen. They simply do not happen by themselves.
2. 1. 7 Heat and work • Heat (Q) is the exchange of thermal energy from a hot body to a cold body. It is a kind of energy transferred in a driving force of temperature difference. • the zeroth law of thermodynamics • If two bodies are in thermal equilibrium with a third body, they are also in thermal equilibrium with each other. • Sign Convention for heat Q • Positive heat in, negative heat out. • Endothermic Q>0, exothermic Q<0.
• Example Hold a piece of ice in your hand until it melts • Solution A • System:You • Surroundings:Ice + the rest of the universe • Q < 0:Heat flows out of the system (you) into the ice. • Solution B • System:Ice • Surroundings:You + the rest of the universe • Q > 0:Heat flows into the system (ice) from you.
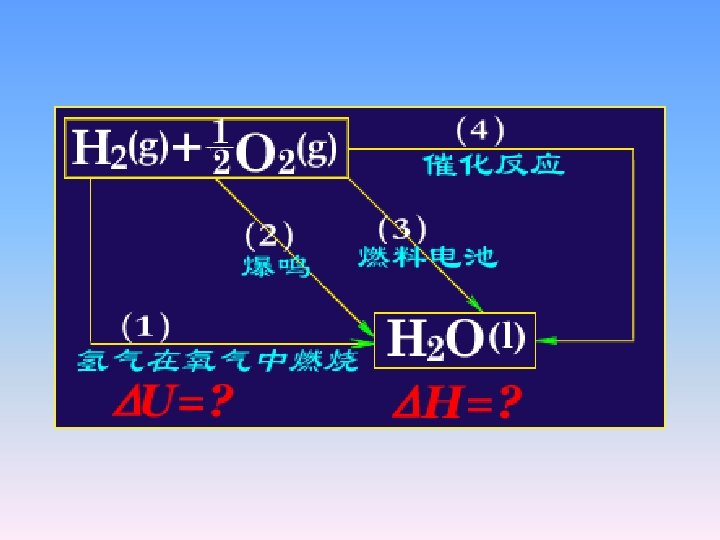
• Example • H 2 burns in a heat insulated (adiabatic) container filled with O 2. what is the heat sign of this process? Positive, negative, or zero?
Work • Work (W): All the transferring form of energy except heat. • There are several kinds of work. Pressurevolume (p. V) work, electrical work, surface work, and mechanical work, etc. • non-volume work (W’): Except pressurevolume work, all the other works.
Work sign 系统 W>0 环境 W<0 According to this convention heat and work are both taken as positive when their direction is into the system of interest. Heat and work are negative when they are out of the system of interest.
Sign of heat and work
The common properties of heat and work • 1. Both are only recognized at the boundaries of a system as they cross the boundaries. • 2. Systems possess energy, but not heat or work. We can say a process has done some work, but we can not say a state has done some work. • 3 Both heat and work are not function of state, they can not be expressed in total differential. The infinitesimal changes are denote by δQ and δW instead of d. Q and d. W, respectively. Their magnitudes depend on the path
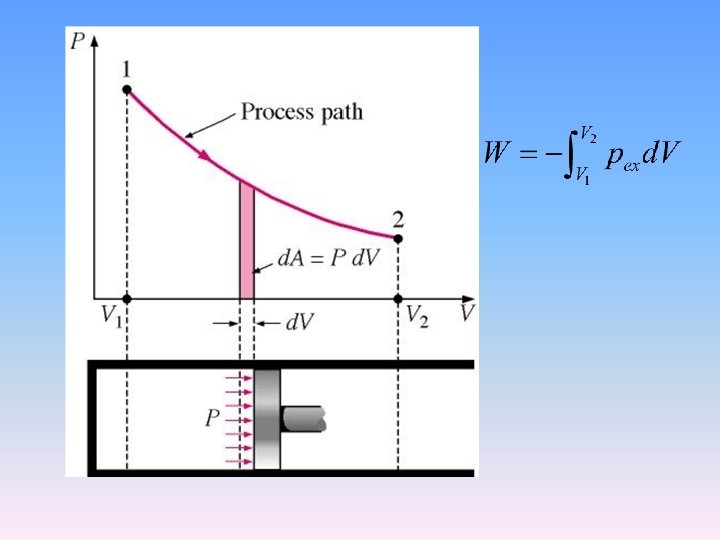
Computation of p. V work
2. 1. 8 Reversible pressure-volume work • Reversible process • is one in which the timescale is assumed to be so slow that every intermediate state deviates only infinitesimally from equilibrium. • After it has taken place, the process can be reversed and causes no change in either the system or its surroundings. That is, all properties of both system and surroundings must be recovered to their original states. There must be no net work or heat. • It is only an ideal process, does not exist in real world.
An example of reversible process Reversible process would be realized by removing the sand one grain at a time
irreversible process • An irreversible process • is a process that cannot return both the system and the surroundings to their original conditions. That is, the system and the surroundings would not return to their original conditions if the process was reversed. • In irreversible process the intermediate states cannot be specified by any set of macroscopic variables and which are not equilibrium states. • All the really happened processes are irreversible processes.
An Irreversible Process
Reversible pressure-volume work • For ideal gas
p. V work
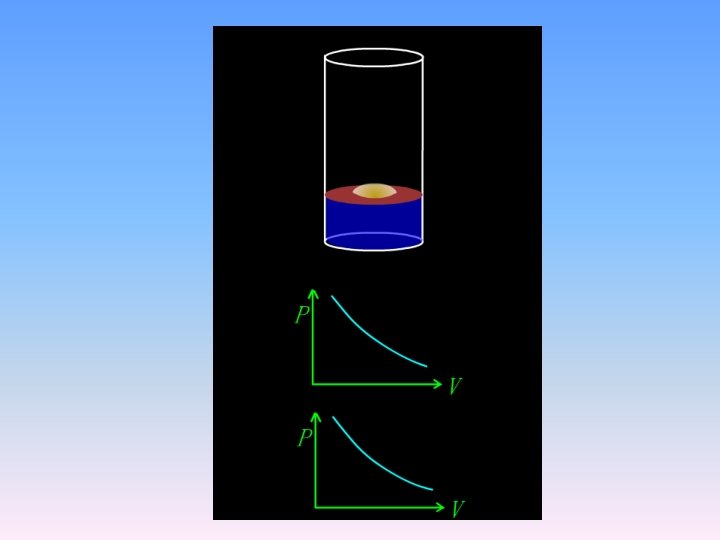
expansion work Fig 2. 1. 4 Three-step expansion of ideal gas at constant temperature
• in a reversible process • The surroundings actually obtained the maximum work from system.
Fig 2. 1. 5 Three-step compression of ideal gas at constant temperature
• in a reversible process • The surroundings actually gives the minimum work to system • The system gives the maximum work to surroundings.
Example • Calculate the work for the isothermal reversible of 1 mol of an ideal gas at 300 K from an initial volume of 20 dm 3 to a final volume of 40 dm 3. • Solution
2. 1. 9 Thermodynamic energy • Thermodynamic energy, also called internal energy • the energy associated with the random, disordered motion of molecules or particles. • It is separated in scale from the macroscopic ordered energy. • Its absolute value is unknown. • Its symbol and unit are U and joule respectively.
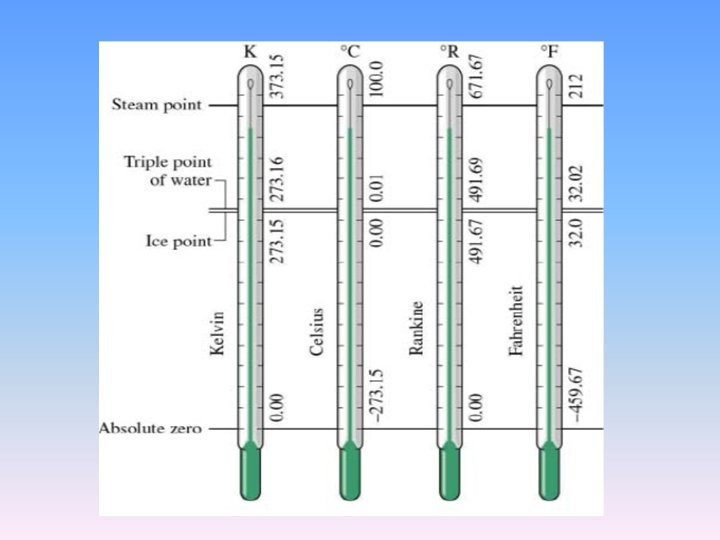
2. 1. 10 Temperature scales
2. 1. 10 Temperature scales • Celsius scale (formerly called the centigrade scale) • the ice and steam points were originally assigned the values of 0℃ and 100°C, respectively. • Fahrenheit scale • The corresponding values on the Fahrenheit scale are 32°F and 212°F • T/℉=1. 8 T/℃+32
thermodynamic temperature scale (K) • Does a temperature has an absolute zero, or 0 K? • From thermal motion of molecules, it should be at 0 K if the motion freeze. • From p. V=n. RT we can see that there is a naturallydefined zero on this scale. p→ 0, T→ 0. • one other fixed point need be defined. its value is set as 273. 16 K at the triple point of water. • This is known as the ideal gas temperature scale. It is identical to Kelvin temperature scale based on Carnot heat engine in Chapter 3.
2. 1. 11 Standard state • Standard state is a state of a system chosen as standard for reference by convention. • p = 100 k. Pa • Standard state of gases: • the state of the pure substance in the gaseous phase at the standard pressure p = 100 k. Pa, assuming ideal behavior. • condensed phase: • the state of the pure substance in the liquid or solid phase at the standard pressure p = p.
• a solute in solution • the state of solute at the standard molality b =1 mol kg-1, standard pressure p =100 k. Pa or standard concentration c = 1 mol dm-3 and exhibiting infinitely dilute solution behavior. • Solvent in solution or component in liquid mixture • Standard state: Pure liquid at 100 k. Pa • The standard state of a system can be defined at any given temperature. • a few texts (especially in related disciplines such as physics and engineering) use 0 degrees Celsius for Standard Temperature and Pressure (STP).
2. 2 The first law of thermodynamics • 2. 2. 1 The first law of thermodynamics • W should include all kinds of work. • The other parlance about the first law of thermodynamics is that a perpetual motion machine of the first kind can not work.
2. 2. 2 Heat and enthalpy • Heat depends on the path in which a process takes place. • QV symbolizes the heat at constant volume. • Qp symbolizes the heat at constant pressure. • If we assume that W’=0, then at constant volume δW = -pd. V =0. • Qv = △U - W • Qv = △U=U 2 -U 1
Heat at constant pressure • • • If we assume that W’=0, then at constant pressure Qp = △U - W Qp = △U + p△V (p 1 = p 2 = pex) Qp = U 2 - U 1 + p (V 2 -V 1) Qp = U 2 - U 1 + p. V 2 - p. V 1 Qp = U 2+ p. V 2 - U 1 - p. V 1 Qp = (U 2 + p. V 2) - (U 1+ p. V 1) Define U + PV = H, which is called enthalpy. It is a state function in Joule. Absolute value and physical meaning unknown.
2. 2. 3 Joules experiment
• Q=0, W=0. Then • △U=Q+W= 0 • Since d. T = 0, it follows that • For ideal gases at constant temperature • H=f(T)
2. 2. 4 Molar heat capacity: C(T)= δQ/d. T
• For ideal gases These two equation are valid for the calculation of enthalpy change or thermodynamic energy change in any process, constant pressure or constant volume is no more a condition for ideal gases.
2. 2. 4. 3 Relation between CV and Cp
The first term indicates the increase of U, and the second volume work. For ideal gas For condensed phase
Heat capacities of ideal gases • Equipartition of Energy Theorem states that molecules in thermal equilibrium have the same average energy associated with each independent degree of freedom of their motion and that the energy is (1/2)RT per mole. • For a monoatomic ideal gas • For a diatomic ideal gas
2. 2. 4. 4 Temperature dependent of molar heat capacity By suming up huge number of experiments, Cp, m=f(T) is ordinary expressed as: Cp,m= a + b T + c T 2 Cp,m= a + b T + c’T-2 Cp,m= a + b T + c T 2 + d T 3 a,b,c,d,c’ are all constats which can be found in appendix D in our textbook. Pay special attention on the valid temperature ragne for the calculation of Cp by using these constants.
average molar heat capacity at constant pressure: The average molar heat capacities apply to two cases: (1) Rough computation of heat (2) temperature change range is samll.
• Theat capacities of gaseous mixture: y is mole frction of compents of gaseous mixture.
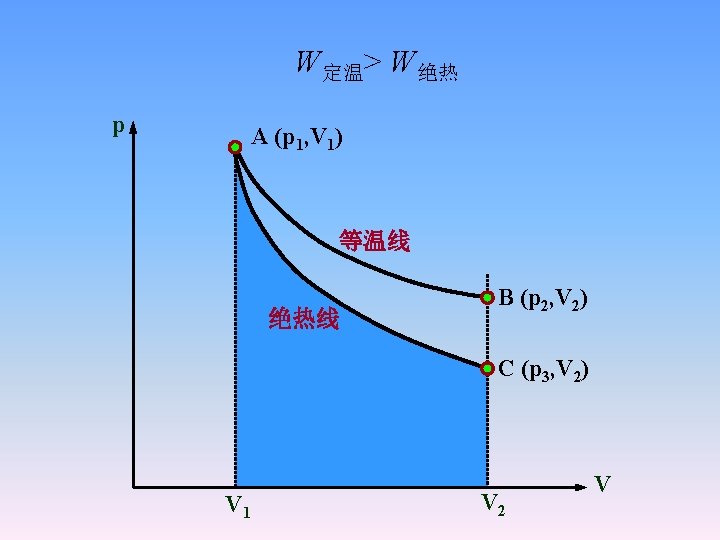
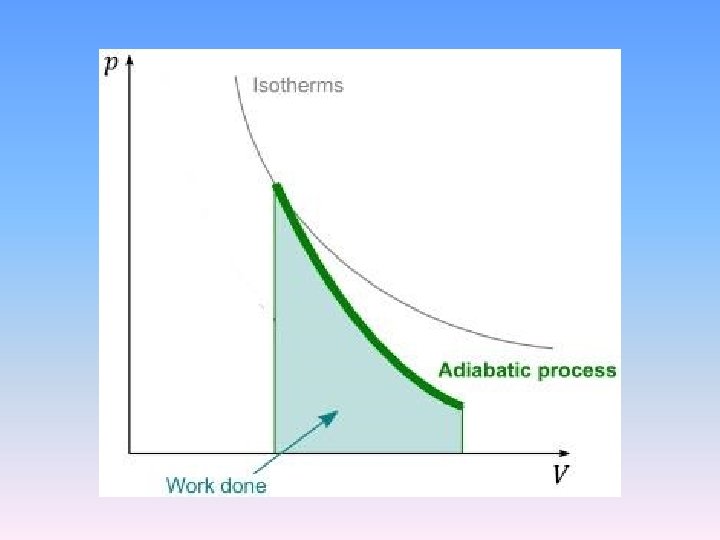
2. 2. 5 Reversible adiabatic process of an ideal gas • From the first law: d. U = δQ + δW • For the reversible adiabatic process δQ = 0. • for ideal gases • γ is called ratio of heat capacity or adiabatic index.
• Using the ideal gas law to replace V, we get the other two equations
• The work done on or take by system is • Integrating this leads to • p 0, V 0 are pressure and volume of system at any state.
The above equation can be changed into a simple form
Comparision of isotherms to adiatats
Example 1 mol 双原子理想气体从25℃,100 k. Pa 突然绝热恒外压 减压至 50 k. Pa,求终态温度T 2及W、△U、△H。 解:因为绝热,Q=0, △U=W=-pamb△V n. CV, m(T 2 -T 1)= -p 2(V 2 -V 1) V 2=n. RT 2/p 2; V 1=n. RT 1/p 1 代入上式,解出T 2=255. 56 K △U=n. CV, m△T=(5/2)R(255. 56 -298. 15)=-885. 3 J △H=n. Cp, m△T= (7/2)R(255. 56 -298. 15)=-1239 J △H= △U+△(p. V)=△U+R△T =-885. 3+8. 3145(255. 56 -298. 15)=-1239 J
Example: 4 mol 双原子理想气体从p 1=50 k. Pa, V 1=160 dm 3绝 热可逆压缩至p 2=200 k. Pa。求末态温度T 2及W,△U,△H。 解:先求T 1=p 1 V 1/n. R=240. 53 K T 2=(p 2/p 1)R/Cp, m. T 1=357. 43 K △U=n. Cv, m(T 2 -T 1)=9. 720 k. J △H=n. Cp, m(T 2 -T 1) W=△U
2. 3 Phase transformation phase is a portion of a system that has uniform properties and composition.
Phase change includes from a liquid to a gas (vaporization) from a solid to a liquid (fusion) from a solid to a gas (sublimation) crystal form transition
Phase change at constant pressure The molar change of enthalpy For melting and crystal transition process at constant pressure and constant temperature
• For vaporization and sublimation processes
Temperature dependence of enthalpy of phase change
2. 4 Standard molar enthalpy of reaction 2. 4. 1 Stoichiometric coefficients a. A + b. B = y. Y + z. Z 0=-a. A-b. B+y. Y+z. Z The numbers, a, b, y, and z, showing the relative numbers of molecules reacting, are called the stoichiometric coefficients.
2. 4. 2 Extent of reaction dξ=dn. B/υB for a same reaction, if the equation of chemical reaction is written in different form, νB will also be different, and then extent of reaction ξ will be different too. For example: N 2(g)+3 H 2(g)= 2 NH 3(g) ½ N 2(g)+ 3/2 H 2(g)= NH 3(g)
2. 4. 3 Molar enthalpy of reaction is an enthalpy change of a reaction. For example: △r. Hm is molar enthalpy of reaction; * stands for a pure substance.
2. 4. 4. Standard molar enthalpy of reaction: the enthalpy change per mole for conversion of reactants in their standard states into products in their standard states, at a specified temperature.
2. 5 Calculation of standard enthalpy of reactions 2. 5. 1 Standard molar enthalpy of formation The enthalpy change when one mole of the compound is formed at 100 k. Pa pressure and given temperature from the elements in their stable states at that pressure and temperature. the stable forms of the elements have For example:
Note that • Any form of elements other than the most stable will not be zero; C (diamond), C (g), H (g), and S (monoclinic) are examples.
Calculation of standard enthalpy of reactions
Example Calculate the standard enthalpy of following reaction at 25℃ by using standard molar enthalpy of formation Solution C 2 H 5 OH(g) C 4 H 6(g) H 2 O(g) H 2(g) -235. 10 110. 16 -241. 81 0 note
2. 5. 2 Standard molar enthalpy of combustion Definition: The enthalpy change when a mole of substance is completely burnt in oxygen at a given temperature and standard pressure. The general convention for the products of combustion is as follows. Carbon in organic compound becomes CO 2(g); H becomes H 2 O(l); N becomes N 2(g); S becomes SO 2(g); Cl becomes HCl(aq) and so on. All these complete products have an enthalpy of combustion of zero. For example, under 298. 15 K and standard pressure
For example A B C D
Determination of heat of combustion
2. 5. 3 Dependence of standard molar enthalpy of reaction on temperature a. A + b. B y. Y + z. Z since then It is called Kirchhoff equation
2. 5. 4 The relationship between heat of chemical reaction at constant pressure and volume • reactions involving only solids or liquids • volume work W≈0, and Δ(p. V)≈0, then for solids and liquids Q≈ΔH≈ΔU. • reactions involving gases • the product p. V may be replaced by νBRT •
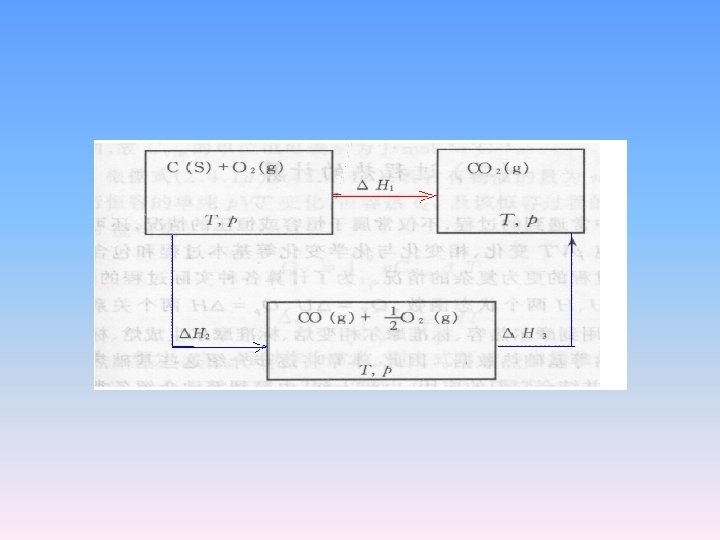
2. 5. 5 Hess’s Law and reaction enthalpy • Hess’s law states that the enthalpy change of any reaction may be expressed as the sum of the enthalpy changes of a series of reactions into which the overall reaction may formally be divided. • The enthalpy change of a reaction at constant pressure or constant volume depends only on the final and initial states, and not on the path connecting them.
Example • some reactions can not be studied directly • C(graphite) + 2 H 2(g) → CH 4(g) • Consider following reactions • The combination of these three reactions from –(a)+2(b)+(c), we get the above studied reaction.
• Then
2. 5. 6 The maximum temperatures of flames and explosions • The temperature reached for a combustion reaction at constant pressure and adiabatic system is known as the maximum temperature of flame. Qp=ΔH=0 • the temperature reached for an explosion in an adiabatic system and at constant volume is called the maximum temperature of explosion. QV=ΔU=0
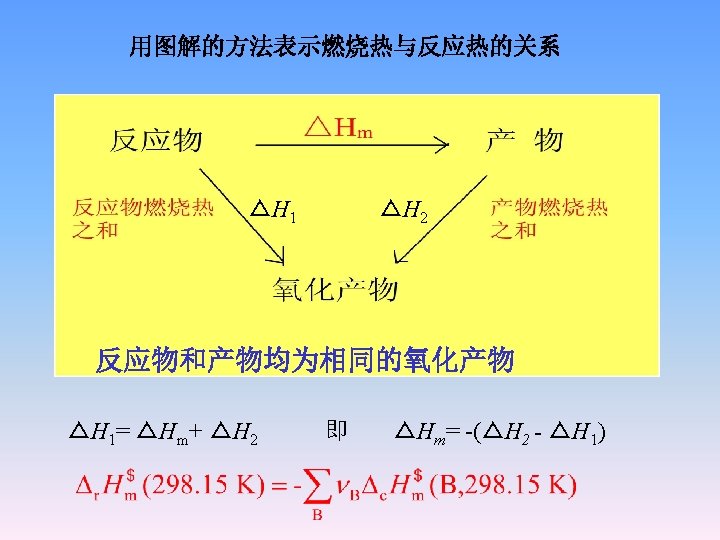
r. H m CH 4(g)+2 O 2(g) CO 2(g)+2 H 2 O(g) c. H m(CH 4, g) △vap. Hm(H 2 O) CO 2(g)+2 H 2 O(l) r. H m = c. H m(CH 4, g)+ 2 vap. Hm(H 2 O) =-802. 286 k. J
对于含 1 mol甲烷(CH 4, g) 的系统,含氧气 4 mol ,氮气(4/0. 21)× 0. 79 mol=15. 05 mol,则 始态T 0=298. 15 K CH 4(g)1 mol, O 2 (g) 4 mol N 2 (g) 15. 05 mol Qp= H=0 恒压绝热 T CO 2(g)1 mol, H 2 O (g) 2 mol O 2 (g) 2 mol, N 2 (g) 15. 05 mol r. H m H 2 T 0=298. 15 K CO 2(g)1 mol, H 2 O (g) 2 mol O 2 (g) 2 mol, N 2 (g) 15. 05 mol Qp= H = r. H m + H 2 =0
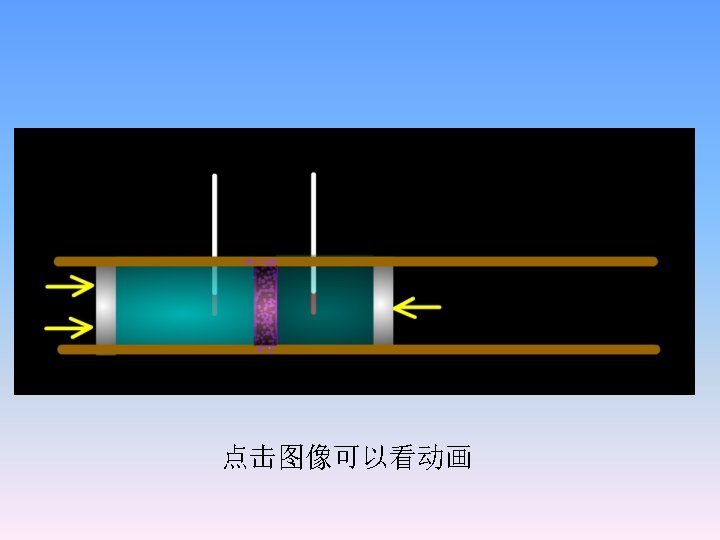
2. 6 Joule-Thomson effect The experiment by Joule and Thomson showed that H of a real gas is not only the function of T, but also the function of p. 1 The experiment by Joule and Thomson Adiabatic wall thermometer p 1, V 1, T 1 p 2, V 2, T 2 Porous plug throttle expansion ,p 1>p 2
Show that for ideal gases H= f ( T, p) Throttling:d H=0)
Structure of air-conditioner
Structure of refrigerator
Operating principle of a refrigerator
Animation of refrigeration • Compression-type refrigerating machine 01 • Compression-type refrigerating machine 02 • Linde cycle for liquefaction of gas
James Prescott Joule 1818~ 1889
Sir William Thomson working on a problem of science in 1890
Summary • △U=Q+W (reversible process) (adiabatic reversible) p. Vγ=const (adiabatic reversible for ideal gas)
Reaction heat Throttling expansion:
Home work • • Page 72 2. 1 (basic concept) 2. 2 (basic concept) 2. 3 (calculation of work) 2. 4 (calculations of work and heat) 2. 9 (Calculate Q, W, ΔU, and ΔH ) 2. 12 (Calculation of Wr, Q, ΔU, ΔH of phase change process)
• 2. 20 (Calculation of adiabatic process) • 2. 25 (Calculation of the enthalpy of formation) • 2. 24 (reaction heat)