CHROMATOGRAFIA Chromatografia jest metod rozdzielania oraz analizy jakociowej

 składników jednorodnych mieszanin w")



 Chromatos= barwa+ grapho= pisze Badał")


 B Po wprowadzeniu mieszaniny substancji A")

 -")

 lub")


















 Jest działem HPLC, gdzie procesy rozdzielcze oparte są na reakcjach wymiany")






 faza ruchoma (eluent) sorbent")























 LINOMAT 5 nanoszenie analitu rozwijanie")
![ROZDZIELANIE MIESZANINY FOSFOLIPIDÓW nanoszona ilość wzorca związku [µg/plamkę] PS PC PE 1 2 3](https://slidetodoc.com/presentation_image_h/ac24b6b5cc1d1d98ef22ec298d55d975/image-67.jpg "ROZDZIELANIE MIESZANINY FOSFOLIPIDÓW nanoszona ilość wzorca związku [µg/plamkę] PS PC PE 1 2 3")
 10000 7500")
")


 Modyfikacja polega na związaniu aktywnych grup hydroksylowych krzemionki")

")



 z: aluminium, tworzywa sztucznego, szkła.")
 płytki pokryte polarnym sorbentem (najczęściej żel krzemionkowy),")







- Slides: 86
CHROMATOGRAFIA
Chromatografia jest metodą rozdzielania ( oraz analizy jakościowej i ilościowej) składników jednorodnych mieszanin w wyniku ich różnego podziału między dwie fazy układu chromatograficznego ruchomą i nieruchomą. OBECNIE WIĘKSZOŚĆ PROBLEMÓW ANALITYCZNYCH JEST ROZWIĄZYWANA METODAMI CHROMATOGRAFICZNYMI faza nieruchoma (stacjonarna, sorbent) ciało stałe lub ciecz unieruchomiona na nośniku, na których zachodzi proces rozdzielania składników badanej mieszaniny. faza ruchoma (eluent) ciecz, gaz lub fluid w stanie nadkrytycznym wykorzystywane do rozdzielenia składników badanej mieszaniny. elucja wymywanie składników rozdzielanej mieszaniny przez eluent.
UKŁADY CHROMATOGRAFICZNE faza stacjonarna ciało stałe ciecz faza ruchoma gaz ciecz PODZIAŁU TECHNIK CHROMATOGRAFICZNYCH można dokonać ze względu na: 1. Stan skupienia fazy ruchomej gaz - chromatografia gazowa, ciecz - chromatografia cieczowa, fluid w stanie nadkrytycznym - chromatografia fluidalna (nadkrytyczna). 2. Stan skupienia fazy stacjonarnej ciało stałe - chromatografia adsorpcyjna, ciecz naniesiona na nośnik - chromatografia podziałowa.
3. Naturę zjawisk będących podstawą procesu chromatograficznego chromatografia adsorpcyjna rozdział mieszanin następuje dzięki różnemu powinowactwu adsorpcyjnemu poszczególnych składników mieszaniny względem fazy stacjonarnej chromatografia podziałowa rozdział mieszanin następuje dzięki różnicom w wartościach współczynników podziału składników między dwie nie mieszające się fazy: stacjonarną i ruchomą chromatografia jonowymienna podstawą rozdziału jest wymiana jonowa między jonami z roztworu, a jonami związanymi z fazą stacjonarną, która stanowią jonity chromatografia żelowa (chromatografia sitowa, sączenie molekularne) o rozdziale decydują rozmiary cząstek substancji oraz żelu wypełniającego kolumnę
Z. Witkiewicz „Podstawy chromatografii” – WNT
Wynalazcą chromatografii jest Michaił Semenowicz Cwiet ( 1872 -1919) Chromatos= barwa+ grapho= pisze Badał on ekstrakty roślinne otrzymane przy użyciu eteru naftowego jako ekstrahenta. 1903 r. Warszawa
Badania Michaiła Semenowicza Cwieta: Ø Ekstrakty wprowadzał do kolumienki wypełnionej węglanem wapnia Ø Po zaabsorbowaniu składników ekstraktu wymywał je benzenem Ø W efekcie oddziaływania składników ekstraktu z węglanem wapnia i wskutek oddziaływań z rozpuszczalnikiem nastąpiło rozdzielenie tych składników.
Pierwszy zestaw chromatograficzny stosowany przez M. S Cwieta
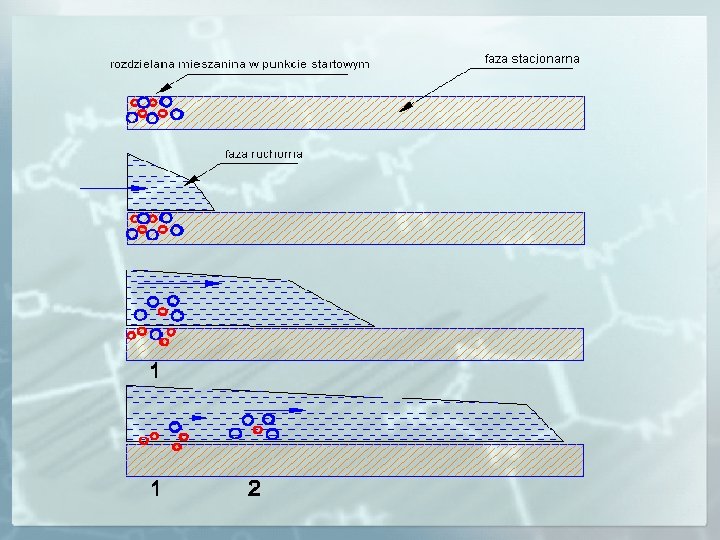
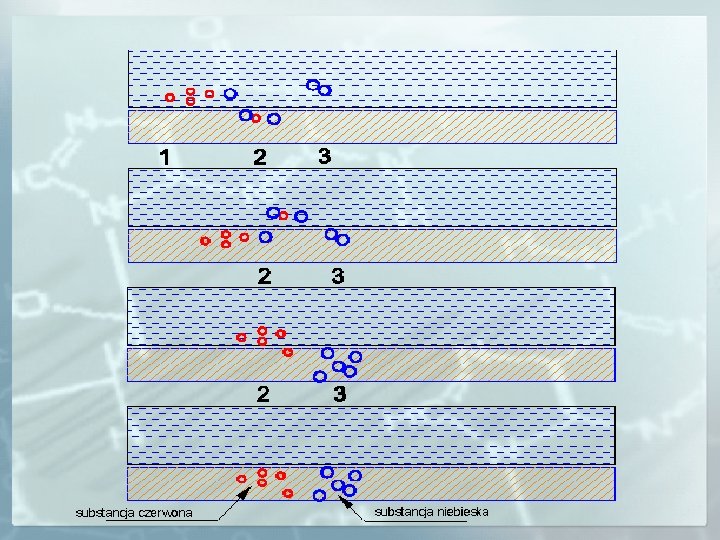
ISTOTA ROZDZIELANIA CHROMATOGRAFICZNEGO t=0 A faza ruchoma (fr) B Po wprowadzeniu mieszaniny substancji A i B do fazy ruchomej (czas t=0) zaczyna się proces jej rozdzielania. faza nieruchoma (fs) t 1 fr W czasie t 1 obserwuje się różny podział składników mieszaniny między obie fazy. A B Substancja A wykazuje mniejsze powinowactwo względem fazy stacjonarnej (oddziałuje z fazą nieruchomą znacznie słabiej) niż substancja B, a co za tym idzie porusza się wzdłuż sorbentu znacznie szybciej niż substancja B. fs t 2 fr A B ~ W czasie t 2 składnik A został oddzielony i opuścił układ chromatograficzny. fs t 3 fr B ~ fs A W czasie t 3 oba składniki mieszaniny zostały rozdzielone i opuściły układ chromatograficzny.
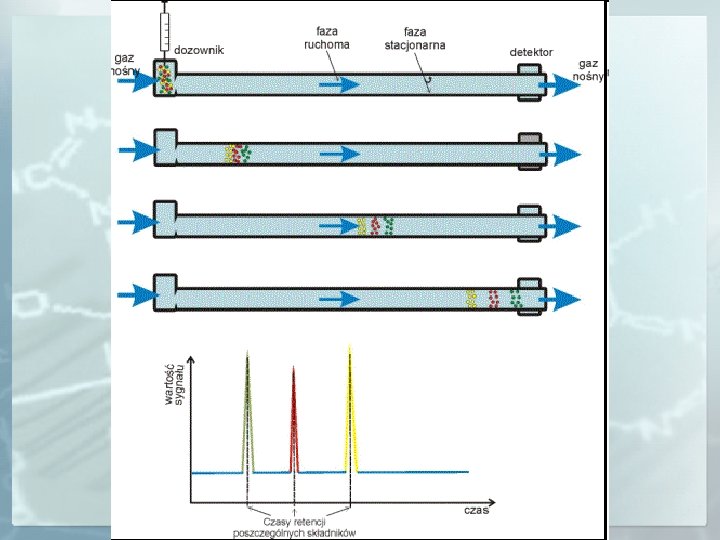
Chromatografia gazowa to technika analityczna oparta na rozdziale składników badanej mieszaniny pomiędzy poruszającym się strumieniem gazu nośnego (faza ruchoma) a wypełnieniem kolumny (faza stacjonarna). Siłą napędową migrującej substancji jest poruszająca się faza ruchoma, a siłą przytrzymującą jest powinowactwo substancji do fazy stacjonarnej. Całkowity czas jaki substancja spędza w kolumnie jest jej czasem retencji (t. R). faza ruchoma gaz ciało stałe faza stacjonarna ciecz osadzona na nośniku
Chromatografia gazowa GC - analiza lotnych zw. org. (twrz <400 o C ) - analiza substancji nielotnych (cukry aminokwasy, steroidy) po derywatyzacji - pirolityczna chromatografia gazowa *umożliwia rozdzielanie, identyfikację i oznaczanie mieszanin kilkuset składników
Próbkę do dozownika wprowadza się za pomocą strzykawki (gazy, ciecze, roztwory ciał stałych) lub zaworem dozującym w przypadku gazów. Próbka, która wchodzi do kolumny musi mieć postać gazu lub pary. Składniki próbki ciekłej lub stałej w postaci roztworu odparowują w dozowniku i są przenoszone do kolumny w strumieniu gazu nośnego.
Gazy nośne Gaz nośny jest fazą ruchomą w GC. Najczęściej stosowanymi gazami są • Wodór • Azot • Argon • Hel • Rzadko stosowany jest ksenon lub mieszanina gazów Rodzaj gazu ma mały wpływ na wynik rozdzielenia chromatograficznego. Gaz powinien być chemicznie obojętny względem wypełnienia kolumny i składników mieszaniny rozdzielanej.
Gazy nośne Na pracę detektora wpływa czystość gazu. Wpływa ona także na trwałość wypełnienia kolumn. Gaz nośny nie może zawierać zanieczyszczeń, ponieważ pod ich wpływem ciekłe fazy stacjonarne mogą ulegać przemianom chemicznym i zmieniać swoje właściwości a adsorbenty mogą ulec dezaktywacji. Gazy te przede wszystkim nie powinny zawierać tlenu, pary wodnej i węglowodorów.
Kolumny W kolumnie chromatograficznej zachodzi właściwy proces chromatografowania. Rozróżniamy następujące rodzaje kolumn: • pakowane o średnicy wewnętrznej 2 -6 mm i długości kilku metrów, • mikropakowane o średnicy 0, 8 - 1, 2 mm i długości kilkunastu metrów , • kapilarne o średnicy 0, 2 - 0, 6 mm i długości kilkudziesięciu metrów, • preparatywne o średnicy ponad 6 mm i długości kilku metrów, • mikrokapilarne o średnicy mniejszej niż 0, 1 mm i długości do kilkudziesięciu metrów.
Kolumny pakowane Kolumny te sporządzone są z materiałów nieaktywnych chemicznie i katalitycznie. Najczęściej wykonane są ze stali nierdzewnej lub szkła. Kolumnę pakowana wypełnia się adsorbentem lub nośnikiem z osadzoną fazą ciekłą. Nośniki i adsorbenty mają kształt ziaren o rozmiarach odpowiadających części milimetra. Korzystnie jest aby ziarna miały kształt kulisty. Rys. schemat pakowanych kolumn chromatograficznych
Rys. kolumna pakowana Rys. Piecyk chromatografu z dozownikiem i kolumną kapilarną Rys. kolumna kapilarna
Kolumny kapilarne W kolumnach kapilarnych fazy stacjonarne mogą być absorbentami a także cieczami i są osadzane na ściankach kapilar w zróżnicowany sposób. Kolumny dzielimy na: • WCOT- kolumny z gładkimi ściankami pokrytymi ciekłą fazą stacjonarną • PLOT- kolumny z warstwą porowatą na ściankach. Służą do analizy gazów trwałych i związków chemicznych zawierających do 16 atomów węgla i mają temperatury wrzenia do 225 o. C. • SCOT- kolumny, na których ścianki naniesiono nośnik nasycony ciekłą fazą stacjonarną. Grubość warstwy ciekłej fazy stacjonarnej wynosi zwykle 0, 1 -0, 3 mm. Im warstwa ta jest cieńsza tym kolumna jest sprawniejsza, a czasy rozdzielenia są krótsze.
Wady i zalety kolumn kapilarnych Zalety: • Krótszy czas analizy niż przy użyciu kolumn pakowanych • Uzyskanie lepszych efektów rozdzielania • Małe zużycie gazów nośnych i faz stacjonarnych • Brak konieczności stosowania nośników Wady: • Trudniejsza preparatyka niż dla kolumn pakowanych • Zwykle są to kolumny kupowane a nie przygotowywane samodzielnie • Wysoka cena
Adsorbenty dzielimy na: • Nieorganiczne • Organiczne Adsorbenty nieorganiczne: - Sita cząsteczkowe, które są naturalnymi zeolitamiglinokrzemianami. Mają średnicę porów ok. 1 nm. Posiadają zdolność rozdzielania gazów (azot, tlen). - Żel krzemionkowy- stosowany do analizy mieszanin zawierających wodór, powietrze, tlenek węgla, alkany i alkeny. Analizuje się też fosgen, chlorowodór i gazowe związki siarki.
Adsorbenty organiczne -Polimery porowate mają największe znaczenie w chromatografii gazowej. Wykazują dobre właściwości rozdzielcze, są hydrofobowe, obojętne chemicznie i mają dużą retencję wielu związków chemicznych. Służą do oznaczania śladowych ilości wody. - Adsorbenty węglowe. Rzadko stosowanym adsorbentem tego typu jest węgiel aktywny jednak można na nim rozdzielać wodór, tlenek węgla, metan. Częściej stosowane obecnie są sadze grafityzowane, doskonałe do rozdzielenia izomerów wielu substancji.
Ciekłe fazy stacjonarne • Substancje gęste o małej lotności. • Ciekła faza stacjonarna powinna być odporna na działanie podwyższonej temperatury. • Odporność na działanie gazu nośnego • Odporność na działanie substancji chromatografowanej • Do rozdzielania substancji niepolarnych należy stosować fazę niepolarną, a do substancji polarnych- fazę polarną.
Grupy ciekłych faz stacjonarnych • Węglowodory • Silikony • Poliglikole i polimery tlenków alkilenów • Estry • Fazy optycznie czynne • Ciekłe kryształy • Cyklodekstryny
Detektory Istotą działania detektorów jest to, że reagują one na różnice właściwości fizykochemicznych gazu nośnego i gazu , w którym znajdują się substancje eluowane z kolumny. Detektor chromatograficzny powinien: • Wykazywać dużą czułość – sygnał przechodzący przez detektor wykrywanego składnika powinien być możliwie największy • Mieć dużą stabilność linii podstawowej • Mieć dużą odtwarzalność wyników • Charakteryzować się szerokim zakresem liniowości wskazań • Wykazywać dużą wykrywalność • Mieć małą stałą czasową • Być selektywne
Detektor cieplno- przewodnościowy Ogólna cecha tego typu czujnika jest znaczna zmiana ich przewodności elektrycznej przy małej zmianie temperatury. Temperatura detektora musi być ustawiona z dokładnością do dziesiątych części stopnia Celsjusza. Czujnikiem jest spirala, np. niklowa, ze stopu platyny lub wolframowa. Zasada ogólna jest taka, że im większa różnica w przewodnictwie cieplnym między gazem nośnym a wykrywanymi substancjami, tym mniejsza ilość substancji może być wykryta. Rys. detektor TCD
Detektor płomieniowo- jonizacyjny FID- czyli detektor płomieniowo- jonizacyjny wymaga wodoru i sprężonego powietrza do prawidłowego działania. W detektorze tym spalany jest wodór (płomień znajduje się między dwiema elektrodami). Jeżeli do płomienia z kolumny dostarczany jest tylko gaz nośny to wytwarzane są termojony powodujące pojawienie się w układzie stałego prądu jonowego o małym natężeniu. W efekcie na chromatogramie zapisywana jest prosta linia podstawowa. Pik pojawia się gdy do płomienia jonowego razem z gazem nośnym dostarczana jest substancja. Detektor spalając próbkę, niszczy ją. Jest to detektor masowy.
Rys. schemat budowy detektora płomieniowo- jonizacyjnego
SCHEMAT BLOKOWY CHROMATOGRAFU CIECZOWEGO HPLC 4 1 3 5 6 7 8 9 10 11 12 2 1, 2 - zbiorniki eluentu, 3 - pompa, 4 - manometr, 5 - dozownik, 8 - termostat, 9 - przepływomierz, 6 - przedkolumna, 7 - kolumna, 10 - detektor, 11 - kolektor frakcji, 12 - komputer
Wysokosprawna chromatografia cieczowa HPLC Jest stosowana do rozdziału i analizy substancji, których nie można analizować metodą GC lub których rozdzielanie tą metodą nastręcza duże trudności: -związki biologicznie czynne: białka, aminokwasy, polipeptydy, witaminy, cukry, sterydy, kwasy nukleinowe -preparaty farmaceutyczne -pestycydy -węglowodory policykliczne (WWA np. benzo(a)piren) w wodzie, glebie, żywności -związki metaloorganiczne i kompleksowe
Ø Przyjmuje się, że ten rodzaj chromatografii powstał w 1968 r. w Las Vegas Ø W latach siedemdziesiątych XX w. zaczęto produkować na skale handlową pompy, kolumny detektory i fazy stacjonarne Ø W latach osiemdziesiątych XX w. zastosowano elektroniczne opracowanie wyników, wprowadzono komputery i detektor z matrycą diodową
Chromatografia jonowa (IC) Jest działem HPLC, gdzie procesy rozdzielcze oparte są na reakcjach wymiany jonowej (anionity, kationity) *aniony (granica ozn. µg/l –detektor konduktometryczny) *kationy (metali alkalicznych – det. konduktometryczny) (lantanowce - derywatyzacja arsenazo I – det. spektrof. ) *aminokwasy(derywatyzacja ninhydryną–det. spektrof) *kwasy karboksylowe *węglowodany
Chromatogram jonowy kationów Eluent 4 m. M kwas winowy + 0, 75 m. M kwas 2 -pikolinowy ; detektor konduktometryczny
Chromatogram jonowy lantanowców
chromatogram jonowy anionów 1 - bromki 2 - chlorki 3 - żelazocyjanki 4 - azotyny 5 - azotany 6 - siarczany 7 - azydki 8 - szczawiany 9 - molibdeniany 10 - wolframiany 11 - fluorki 12 - winiany 13 - seleniny 14 - fosforany
Chromatografia nadkrytyczna SFC- supercritical fluid chromatography Jest to rodzaj chromatografii, gdzie fazą ruchomą jest substancja w stanie nadkrytycznym. W Polsce wciąż mało stosowana. Chromatografia nadkrytyczna umożliwia analizę związków chemicznych o małej trwałości termicznej, małej lotności, dużych masach cząsteczkowych, których analiza za pomocą GC byłaby wręcz niemożliwa. Można w niej stosować detektory używane w chromatografii gazowej i cieczowej. Analizę wykonuje się szybciej i z lepszą rozdzielczością niż za pomocą chromatografii cieczowej. Rys. Typowy wykres fazowy dowolnej substancji. W rożnych warunkach ciśnienia i temperatury trwałe są rożne stany skupienia. Gaz od cieczy ogranicza linia wrzenia, łącząca punkt potrójny (A) i punkt krytyczny (B). Powyżej punktu krytycznego zanika różnica między gazem i cieczą.
CHROMATOGRAFIA PLANARNA
Chromatografia planarna jest odmianą chromatografii cieczowej, w której faza nieruchoma jest umieszczona na płaszczyźnie chromatografia planarna chromatografia bibułowa bibuła chromatografia cienkowarstwowa faza stacjonarna warstwa sorbentu naniesiona na nośnik
CHROMATOGRAFIA CIENKOWARSTWOWA TLC – Thin Layer Chromatography faza stacjonarna (sorbent) faza ruchoma (eluent) sorbent osadzony na nośniku PŁYTKA CHROMATOGRAFICZNA ciecz pojedyńczy rozpuszczalnik lub mieszanina rozpuszczalników
ETAPY PROCESU CHROMATOGRAFICZNEGO W TLC
ETAP I - PRZYGOTOWANIE PŁYTKI linia końca Z arkusza np. 20 x 20 cm wycina się płytkę o wymiarach odpowiednich do przeprowadzanego oznaczenia. W odległości 1 -2 cm od brzegu płytki rysuje się tzw. linię startu, na która będzie nanosić się próbki. Na linii można delikatnie zaznaczyć miejsca nanoszenia próbek. linia startu Od drugiego końca płytki w takiej samej odległości od jej brzegu wyznacza się tzw. linię końca. Nośnik, na którym osadzony jest sorbent może być z aluminium, szkła lub poliestru.
ETAP II - NANOSZENIE PRÓBEK Na linie startu nanosi się próbkę rozdzielanej mieszaniny oraz wzorce. Próbki należy nanosić tak, aby nie uszkodzić sorbentu. Gdy roztwór jest rozcieńczony i musimy nanieść większą ilość substancji wówczas nanosimy próbki kilkakrotnie, za każdym razem odparowując rozpuszczalnik. Średnica pojedynczej plamki nie powinna być większa niż 2 mm. Próbki nanosi się za pomocą: mikropipet, mikrostrzykawek. kapilar, automatycznych aplikatorów (100 nl – 100 µl) PB W 1 W 2 W 3
ETAP III - ROZWIJANIE CHROMATOGRAMÓW komory chromatograficzne pionowe poziome
ZASADA DZIAŁANIA KOMORY POZIOMEJ
KOMORY TYPU „SANDWICH” pionowe 1 – płytka chromatograficzna, 2 – płytka szklana ograniczająca przestrzeń komory, 3 – zbiornik eluentu, 4 – eluent, 5 – pokrywa. 2 -3 mm Komory typu „sandwich” mają bardzo małą objętość. Przestrzeń, w której znajdują się pary rozpuszczalnika jest ograniczona dlatego nie jest konieczne kondycjonowanie komory. Zastosowanie komory typu „sandwich” zmniejsza 10 -20 krotnie zużycie eluentu.
ROZWIJANIE CHROMATOGRAMÓW W KOMORZE PIONOWEJ W komorze chromatograficznej znajduje się eluent w takiej ilości, aby po wstawieniu płytki poziom cieczy znajdował się poniżej linii startu. W momencie zetknięcia się z sorbentem eluent zaczyna się poruszać wzdłuż płytki. Rozpoczyna się proces, który nazywa się rozwijaniem chromatogramu, który kończy się gdy eluent osiągnie linię końca. W wyniku oddziaływania substancji chromatografowanych z sorbentem i fazą ruchomą następuje rozdzielenie mieszaniny na poszczególne składniki. Na płytce otrzymujemy szereg plamek odpowiadających poszczególnym składnikom mieszaniny. Otrzymujemy chromatogram. Składniki, które oddziałują silniej z sorbentem niż z eluentem dają plamki położone bliżej linii startu. Plamki położone dalej od linii startu odpowiadają substancjom oddziałującym silniej z eluentem niż z sorbentem. PB W 1 W 2 W 3
ROZWIJANIE CHROMATOGRAMÓW W KOMORZE POZIOMEJ „od brzegu do brzegu”, jednoczesne rozwijanie od przeciwległych brzegów, odśrodkowe. pojedynczej próbki miejsce naniesienia próbki i doprowadzenia eluentu kilku próbek miejsce doprowadzenia eluentu miejsca naniesienia próbek plamki rozdzielonych składników próbki okręgi rozdzielonych składników próbki
dośrodkowe rozwijanie chromatogramu miejsca naniesienia próbek plamki rozdzielonych składników próbki
Zalety dośrodkowego rozwijania chromatogramów wykorzystuje się stosując płytki z trójkątnie ukształtowanymi w wyniku usunięcia sorbentu fragmentami. linie tworzące boki trójkąta powstałe przez usunięcie sorbentu plamki rozdzielonych składników próbki miejsca naniesienia próbek
dwukierunkowe rozwijanie chromatogramów W przypadku złożonych mieszanin, których składniki nie rozdzielają się po jednokrotnym chromatografowaniu stosuje się rozwijanie dwukierunkowe prostopadłe. dwukierunkowe kierunek ruchu eluentu II 3 2 1 PB
dwukierunkowe rozwijanie chromatogramów 5+6 3+4 1+2 PB kierunek ruchu eluentu II kierunek ruchu eluentu I . 4 6 2 3 1 5 PB
ROZWIJANIE CHROMATOGRAMÓW W KOMORZE CIŚNIENIOWEJ płytka do rozwijania chromatogramu w komorze ciśnieniowej zaimpregnowany brzeg płytki miejsca naniesienia próbek rowek w warstwie sorbentu miejsce doprowadzenia eluentu
ETAP IV - WIZUALIZACJA CHROMATOGRAMÓW Gdy składniki rozdzielanej mieszaniny są barwne na chromatogramie otrzymuje się serię barwnych plamek, dzięki czemu łatwo ocenić otrzymane wyniki. Częściej jednak zdarza się, że wszystkie lub niektóre składniki mieszaniny są bezbarwne. Bezpośrednia analiza wyników jest wtedy niemożliwa. Konieczne jest uwidocznienie plamek rozdzielonych substancji. W celu uwidocznienia plamek przeprowadza się tzw. wizualizację (wywołanie) chromatogramu PB W 1 W 2 W 3
SPOSOBY WIZUALIZACJI CHROMATOGRAMÓW 1. wizualizacja metodami chemicznymi odczynniki w postaci roztworów spryskiwanie zanurzanie w r-rze odczynniki w postaci gazowej nasycanie parami (jod, brom, amoniak) Odczynniki do wizualizacji chemicznej uniwersalne reagują z wieloma związkami zawierającymi różne grupy funkcyjne np. manganian(VII) potasu, pary jodu, bromu grupowe reagują z z określoną grupą związków chemicznych np. ninhydryna (aminokwasy), sole diazoniowe (aminy aromatyczne, fenole), odczynnik Dragendorffa-HBi. I 4 (alkaloidy, wiele związków o charakterze zasadowym)
PB W 1 W 2 W 3
2. Naświetlanie przy zastosowaniu lampy UV emitującej promieniowanie o dł. fali 254 nm lampa UV płytka nie naświetlana lampą UV lampa UV z komorą płytka naświetlana lampą UV
3. Metoda ciekłokrystaliczna W metodzie tej porowatą przezroczystą folię nasyca się ciekłym kryształem. Folia w świetle spolaryzowanym ma jednakową barwę na całej powierzchni. Po dociśnięciu folii do płytki substancje znajdujące się na niej – po rozdzieleniu mieszaniny – w postaci plamek przechodzą do ciekłego kryształu. W wyniku tego uporządkowana struktura ciekłego kryształu przekształca się w ciecz izotropową i na folii pojawiają się plamki o barwie różniącej się w świetle spolaryzowanym od tła. Plamki te są odbiciem chromatogramu znajdującego się na płytce.
ANALIZA JAKOŚCIOWA Parametrem, który charakteryzuje substancję jakościowo w danym układzie chromatograficznym tzn. w określonych warunkach rozdziału jest tzw. współczynnik opóźnienia Rf (Retardation Factor). PB W 1 W 2 A B A – droga migracji substancji chromatografowanej mierzona od linii startu do środka plamki, B – droga migracji eluentu mierzona od linii startu do czoła fazy ruchomej (linia końca). Identyfikacja substancji polega na na porównaniu wartości jej współczynnika Rf z wartością Rf wzorca otrzymaną w warunkach identycznych jak stosowane podczas analizy. wzorca
ANALIZA ILOŚCIOWA PB Cw 1 Cw 2 Cw 3 cw 1 < cw 2 < cw 3 p. B ≈ cw 2 Przy wykorzystaniu chromatogramów plamkowych możliwe jest zatem jedynie półilościowe oznaczenie zawartość substancji próbce W celu otrzymania dokładnej zawartości substancji rozdzielanych stosuje się metodę zwaną densytometrią
DENSYTOMETRIA Densytometria jest techniką zamiany chromatogramów plamkowych na chromatogramy pikowe. Światło o wybranej, odpowiedniej długości fali pada wzdłuż linii rozwijania chromatogramu na poruszającą się płytkę. Jest ono pochłaniane w różny sposób przez sam adsorbent oraz przez adsorbent z zaadsorbowaną substancją (miejsca z plamkami substancji). Dlatego światło odbite od płytki ma różną intensywność w zależności od miejsca na które padło. W wyniku rejestracji zmian intensywności światła odbitego od płytki otrzymujemy chromatogramy pikowe nazywane densytogramami. Każdej plamce na chromatogramie plamkowym odpowiada pik na densytogramie. 10 7 13 3 8 5 4 2 1 11 3 12 densytogram 6 9 chromatogram plamkowy 1 2 3 4 5 6 7 8 9 10 11 12 13
densytometr praca z densytometrem densytogram 2 D densytogram 3 D dla różnych długości fali
PROCEDURA ANALITYCZNA Z ZASTOSOWANIEM METOD ANALIZY OBRAZU (IMAGE ANALYSIS) LINOMAT 5 nanoszenie analitu rozwijanie chromatogramu wizualizacja ANALIZA OBRAZU – PROGRAM TLSee chromatogram plamkowy skanowanie chromatogramu konwersja na chromatogram pikowy
ROZDZIELANIE MIESZANINY FOSFOLIPIDÓW nanoszona ilość wzorca związku [µg/plamkę] PS PC PE 1 2 3 4 5 1. 5 5. 0 7. 0 10 15 5. 0 9. 0 15 20 25 1. 0 7. 0 10 15 20 chloroform : metanol : 25% roztwór amoniaku (65: 25: 4, v/v/v) przykładowy chromatogram plamkowy oraz pikowy prezentujący zakres oznaczalności poszczególnych fosfolipidów
WYZNACZANIE ZAKRESU PROSTOLINIOWOŚCI R 2 = 0. 9984 pole powierzchni piku (n=6) 10000 7500 5000 fosfatydyloetanoloamina zakres [µg/plamkę] 1 -20 0. 09 2500 0 0 8000 pole powierzchni piku (n=6) fosfolipid 5 10 15 naniesiona ilość PE [�g/plamkę] 0. 28 20 R 2 = 0. 9994 7000 6000 5000 4000 fosfolipid sfingomielina zakres [µg/plamkę] 2. 5 -20 0. 51 3000 2000 0 5 10 15 naniesiona ilość SM [µg/plamkę] 20 LOD – granica wykrywalności, LOQ – granica oznaczalności 1. 7
FAZA STACJONARNA (SORBENT)
RODZAJ SORBENTU wywiera znaczny wpływ na przebieg i wynik chromatografowania żel krzemionkowy grupa silanolowa grupa siloksanowa Obecność grup hydroksylowych sprawia, że jest sorbentem polarnym zdolnym tworzyć wiązania z polarnymi związkami chromatografowanymi. Im więcej wolnych grup hydroksylowych, tym większa efektywność rozdziału. Ma największe znaczenie w TLC.
Zalety żelu krzemionkowego: uziarnienie o pożądanej wielkości i rozmiarach porów, określona powierzchnia właściwa, duża wytrzymałość mechaniczna. Wady żelu krzemionkowego: higroskopijność żelu powodująca dezaktywację powierzchni żelu co wpływa niekorzystnie na jego właściwości i odtwarzalność wyników analizy.
modyfikowany żel krzemionkowy (fazy związane) Modyfikacja polega na związaniu aktywnych grup hydroksylowych krzemionki za pomocą łańcuchów alkilowych o 2, 8 lub 18 atomach węgla. ( RP 2, RP 8, RP 18 ) REAKCJA SILANIZACJI faza C 8
tlenek glinu Sorbent polarny Rozdzielanie mieszanin zawierających np. alkaloidy, fenole, steroidy, witaminy, jony metali, lipidy. krzemian magnezu (Florisil) celuloza W postaci włóknistej (bardzo cienkie włókna) i mikrokrystalicznej. Rozdzielanie związków hydrofilowych np. : kwasów fosforowych, fosforanów, glikozydów, aminokwasów, alkoholi i cukrów. celuloza acetylowana Niepolarny sorbent, stosowany w chromatografii z eluentami zawierającymi wodę. Rozdzielanie związków takich jak: alkaloidy, lipidy, antrachinony, nitrofenole. poliamid i poliamid acetylowany Może być wielokrotnie regenerowany poprzez usuwanie po każdym chromatografowaniu obecnych na płytce eluentem o dużej sile elucji. Rozdzielanie np. : fenoli, glikozydów, nukleozydów, pestycydów. substancji
FAZA RUCHOMA (ELUENT)
PB W 1 W 2 W 3 Prawidłowa analiza jakościowa i ilościowa TLC jest możliwa gdy: plamki poszczególnych składników mieszaniny są nierozmyte, plamki poszczególnych składników są dobrze rozdzielone. Taki wynik chromatografowania osiąga się wówczas gdy kierując się oczekiwanym składem analitu oraz rodzajem sorbentu dobierze się odpowiednią fazę ruchomą. Siłę oddziaływań danego rozpuszczalnika z określonym sorbentem charakteryzuje tzw. siła elucji rozpuszczalnika. sorbent siłę elucji wyznaczają wzrost siły elucji wywołuje polarny polarność i polaryzowalność cząsteczek rozpuszczalnika wzrost polarności i polaryzowalność cząsteczek rozpuszczalnika niespecyficzne siły Van der Waalsa wzrost rozmiarów niepolarnych fragmentów cząsteczek fazy ruchomej niepolarny
Faza ruchoma jest najczęściej mieszaniną dwóch lub trzech rozpuszczalników zmieszanych w proporcjach zapewniających optymalny rozdział badanej mieszaniny. SZEREG ELUOTROPOWY DLA SORBENTÓW POLARNYCH n-heksan, CCl 4, toluen, chloroform……. acetonitryl, etanol, metanol, woda, CH 3 COOH siła elucji rośnie Dla chromatografowania na niepolarnych fazach stacjonarnych siła elucji rośnie w kierunku przeciwnym. Ogólna zasada wyboru eluentu jest następująca: polarna faza stacjonarna mniej polarna faza ruchoma np. żel krzemionkowy np. heksan, toluen chloroform niepolarna faza stacjonarna bardziej polarna faza ruchoma np. faza C 18 np. acetonitryl, metanol, woda normalny układ faz odwrócony układ faz Zmiana składu eluentu w bardzo niewielkim zakresie wywiera duży wpływ na jakość rozdziału mieszaniny.
analit polarny słabo polarny , niepolarny faza stacjonarna polarna większa mniejsza niepolarny słabo polarny , polarny polarność eluentu niepolarna większa Rozdzielanie mieszanin za pomocą chromatografii cienkowarstwowej prowadzi się w celu: analitycznym – grubość sorbentu 0. 1 -0. 25 mm, preparatywnym – grubość sorbentu 0. 5 -2 mm.
Sorbent osadza się na podłożu (płytkach) z: aluminium, tworzywa sztucznego, szkła.
RODZAJE PŁYTEK DO ROZDZIELANIA ANALITYCZNEGO zwykłe (TLC) płytki pokryte polarnym sorbentem (najczęściej żel krzemionkowy), stosowane w normalnym układzie faz. wysokosprawne (HPTLC – High Performance Thin Layer Chromatography) stosowane są sorbenty o drobniejszym uziarnieniu i węższych frakcjach sitowych niż w przypadku TLC. płytki RP (RP-TLC – Reverse Phase Chromatography) płytki pokryte modyfikowanym żelem krzemionkowym, stosowane w odwróconym układzie faz. pokryte sorbentem zawierającym czynnik fluoryzujący (F 254) Dodatek czynnika fluorescencyjnego (siarczek kadmu lub krzemian cynku) ułatwia obserwację chromatografów przy zastosowaniu lampy UV emitującej promieniowanie o dł gości fali 254 nm. W takich warunkach widoczne są plamki substancji gaszących fluorescencję, albo mających inną długość fali światła emitowanego pod wpływem wzbudzenia niż czynnik fluoryzujący. Czynnik fluorescencyjny można stosować się do każdego rodzaju sorbentów.
ze strefą koncentracji strefa koncentracji pokryta szerokoporowatym żelem krzemionkowym koncentracja plamek na granicy faz chromatogram po rozwinięciu plamki naniesione w dowolnym miejscu linia startu – zaczyna się rozdział
PORÓWNANIE PŁYTEK TLC I HPTLC parametr płytki TLC płytki HPTLC 0, 25 0, 20 lub 0, 10 10 - 12 4 - 8 15 - 100 3 - 20 100 - 180 30 - 60 rozdzielenie mieszaniny na składniki dobre lepsze wykrywalność analizowanych substancji dobra lepsza 20 – 100 ml 5 – 20 ml 1 - 5 0, 1 - 1 grubość sorbentu [mm] rozmiar cząstek sorbentu [ µm] Czas rozwijania [min] droga rozwijania chromatogramu [mm] zużycie eluentu objętość nanoszonej próbki µl
§ W identyfikacji białek, aminokwasów i kwasów nukleinowych w laboratoriach biochemicznych § Do oznaczania zanieczyszczeń żywności; tłuszczów, alkoholi, estrów, żywic, witamin, barwników, cukrów itp. § Do wykrywania syntetycznych związków barwiących w mięsie, w sokach owocowych, w wyrobach cukierniczych, w serach, w makaronach, w przyprawach, napojach alkoholowych, w karmie dla zwierząt.
Zalety TLC względem HPLC • Znacznie niższe koszty analiz • Krótszy czas analizy, gdyż na jednej płytce można analizować kilkanaście próbek • Próbki mogą być zanieczyszczone, gdyż płytkę stosujemy tylko jeden raz • Na płytkę można nanosić duże objętości rozcieńczonych roztworów odparowując rozpuszczalnik • Nie trzeba dobierać fazy ruchomej odpowiedniej do detektora, gdyż przed detekcją jest ona odparowana • Analit można wykrywać na płytce wiele razy różnymi metodami
Zalety HPLC względem TLC • Lepsza separacja • Możliwość separacji i oznaczeń nawet kilkudziesięciu związków w jednej próbce • Lepsza dokładność i precyzja • Szerszy zakres oznaczalności • Niższe granice detekcji
W rozwój chromatografii znaczny wkład wnieśli także polscy uczeni. Jednym z nich był prof. A. Waksmundzki (1910 -1998) Duże znaczenie dla rozwoju chromatografii w Polsce miało długoletnie przewodniczenie przez profesora Komisji Analizy Chromatograficznej Komitetu Chemii Analitycznej Polskiej Akademii Nauk. Z jego inicjatywy została zorganizowana jedna z pierwszych pracowni w Polsce, najpierw chromatografii gazowej a następnie cieczowej kolumnowej
Pytania egzaminacyjne 1. Wykorzystanie TLC do analizy jakościowej i ilościowej. 2. Oznaczanie ilościowe w normalnym i odwróconym układzie faz 3. Fazy stacjonarne w TLC 4. Sposoby wizualizacji chromatogramów 5. Wyjaśnić skróty TLC, HPTLC, GC, RP 18 6. Co to jest szereg eluotropowy i jakie ma znaczenie w chromatografii 7. Jakie fazy stacjonarne i ruchome stosujemy w normalnym i odwróconym układzie faz 8. Opisać budowę komory poziomej i komory typu „sandwicz”