Chapter 17 Free Energy and Thermodynamics 17 2



























 n Now imagine we can take a snapshot of the")





 we need")




 or")









 and H")
 - Σ m. Sº(reactants) ΔSº= 2")


 of a state is n n")



 + 3 H 2(g) ⇄ 2 NH")




![n n n ΔGº = ΣnΔGºf(products) - ΣnΔGºf(reactants) = 4 ΔGºf[PCl 3(g)] - ΔGºf[P](https://slidetodoc.com/presentation_image_h/c9817223b8db551af54c384af9da55b2/image-64.jpg "n n n ΔGº = ΣnΔGºf(products) - ΣnΔGºf(reactants) = 4 ΔGºf[PCl 3(g)] - ΔGºf[P")

 + 5 O 2(g)")




 H 2 O(l) ΔH >")


 + 3 H 2(g) ⇄ 2")




 n Under standard conditions, the concentrations of all")



")



 n We can see that if ΔGº is negative n")

/(8.")
- Slides: 89
Chapter 17 – Free Energy and Thermodynamics
17. 2 - Spontaneous and Nonspontaneous Processes n The first law of thermodynamics simply states that energy is conserved n n n Energy is not created or destroyed in any process Energy can be transferred between a system and surroundings, or energy can be converted from one form to another. Mathematically as n n n ΔE = q + w q = heat absorbed by system w is work done on the system by surroundings
n The first law helps us keep track of energy, but does not talk about the extent of the processes that involve energy n n n Example: Sodium metal and chlorine gas combine to form sodium chloride We never find sodium chloride decomposing back into sodium metal and chlorine gas n n Yet energy is conserved But one process occurs, and other does not
Spontaneous Process n A process that occurs of its own accord without any ongoing outside intervention is said to be spontaneous n So a spontaneous process is one that proceeds on its own without any outside assistance
Reversible n Imagine a brick and the ground n A process involving these two items will only occur in one direction n n The brick will fall down spontaneously Now imagine you see a brick rising from the ground n n Hopefully, you would conclude that the video you were watching was running in reverse A brick falling is spontaneous, but the reverse process is nonspontaneous
Examples of Spontaneous Reactions n A gas will expand into a vacuum n n This would be spontaneous. But the reverse, a gas suddenly condensing into a small amount will not occur n n Would be nonspontaneous We find that any time we have a process that is spontaneous in one direction, it is nonspontaneous in the opposite direction
Even more examples! n Temperature and pressure are important when determining what is spontaneous n If ice is above 0ºC, then the ice melting would be spontaneous n n But above 0ºC, water turning into ice would not be. But if the temperature is below 0ºC, then water turning to ice would be spontaneous, but ice melting would not be
Rate and Spontaneity n A process that is identified as “spontaneous” does not mean that it will occur at an observable rate n n A chemical reaction is spontaneous if it occurs on its own accord, regardless of rate Thermodynamics tells us the direction and extent of the reaction, but nothing about the rate of the reaction
17. 3 - Entropy and the Second Law of Thermodynamics* n To understand spontaneity requires us to look at thermodynamic quantity called entropy n Entropy has been associated with n n The randomness in a system The extent to which energy is distributed among the molecules of a system Entropy is a complex concept that cannot be stated in a simple definition We will look at how entropy is related to heat transfer and temperature n Next section we will look at the molecular significance of entropy
Entropy Change n The entropy of a system, S, is a state function n Like internal energy, E and enthalpy, H That means that the value of S is characteristic of the state of the system So the change in entropy, ΔS, in a system depends ONLY on the initial and final states of the system n n Not path dependent ΔS = Sfinal - Sinitial
Isothermal Processes n n For the special case of an isothermal process, ΔS equals the heat that would be transferred if the process were reversible (qrev), divided by the temperature at which the process occurs But since S is a state function, we can use this equation to calculate ΔS for ANY isothermal process, not just the reversible ones
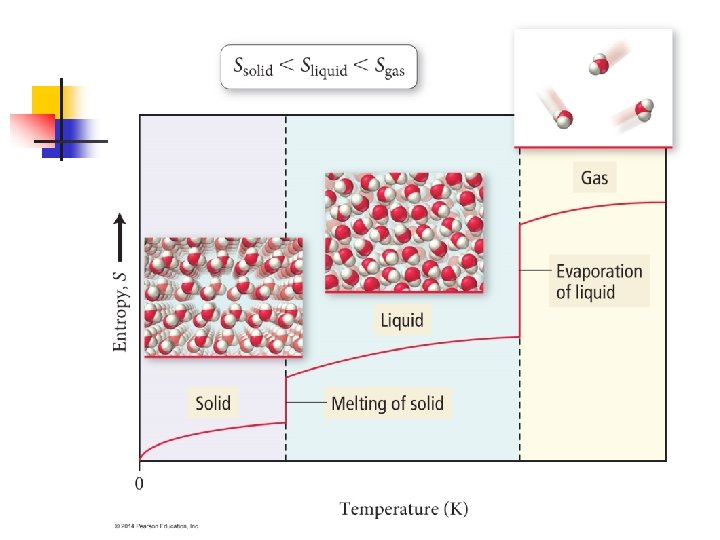
ΔS for Phase Change n n The melting of a substance at its melting point and the vaporization of a substance at its boiling point are isothermal processes Consider the melting of ice n n n At 1 atm pressure, ice and liquid water are in equilibrium with each other at 0ºC. Imagine we melt one mole of ice at 0ºC, 1 atm to form one mole of liquid water at 0ºC, 1 atm. We do this by adding a certain amount of heat to the system (from the surroundings) n q = ΔHfusion
Not done yet n Now imagine that we carry out the change by adding the heat infinitesimally slowly n n n Raising the temperature of the surroundings only infinitesimally above 0ºC. Now the processes is reversible We reverse the process by infinitely slowly removing the same amount of heat, ΔHfusion, from the system n n Using surroundings that are infinitesimally below 0ºC Thus, qrev = ΔHfusion and T = 0ºC = 273 K
n The enthalpy of fusion for water is ΔHfusion = 6. 01 k. J/mol n n Melting is endothermic and so the sign for ΔH is positive So we can now use our previous equation to find ΔSfusion for melting one mole of ice at 273 K
Second Law of Thermodynamics n The key idea to the first law of thermodynamics is that energy is conserved n n To energy lost by system = energy gained by surroundings Entropy is different n n Entropy will increase in any spontaneous process So the sum of the entropy change of the system and surroundings for any spontaneous process is ALWAYS greater than zero n Really, entropy change can tell us whether a process is spontaneous
n Calculate the entropy change of the system and the entropy change of the surroundings when a mole of ice (about 1 ice-cube of ice) melts in the palm of your hand n n Not reversible because the system and surroundings are at different temperatures But because ΔS is a state function, the entropy change is the same regardless of whether or not the process is reversible
n Previously we calculated the entropy change of the system of 1 mole of ice melting n n ΔSsystem= 22. 0 J/K The surroundings immediately in contact with the ice is your hand n n Assume body temperature of 37ºC = 310 K The heat lost by your hand is equal in amount to the heat gained by the ice (but opposite sign) n n q = -6. 01 x 103 J/mol So we can calculate
All Together! n n So the total entropy change is positive ΔStotal = ΔSsystem + ΔSsurroundings ΔStotal = (22. 0 J/K) + (-19. 4 J/K) = 2. 6 J/K Note: n n If temperature of surroundings was some temperature infinitesimally above 273 K, the melting would be reversible At that surrounding temperature, the entropy change of the surroundings would equal -22. 0 J/K, and the total entropy change would be 0
In General n Any irreversible process results in an overall increase in entropy n n A reversible process results in no overall change in entropy This general statement is known as the second law of thermodynamics n n The sum of the entropy of system + entropy of the surroundings is everything So we refer to total entropy change as the entropy change of the universe, ΔSuniv
Variation n For a reversible process: n n Irreversible process: n n ΔSuniv = ΔSsystem + ΔSsurroundings = 0 ΔSuniv = ΔSsystem + ΔSsurroundings > 0 All real, spontaneous processes are irreversible n Therefore, the total entropy of the universe increases in any spontaneous process
Warning n Common misconception n An increase in entropy means it’s spontaneous n Only if you’re focusing on the entire universe. The system’s entropy can increase or decrease and still be spontaneous
The Molecular Interpretation of Entropy* n Before we analyze molecules and entropy, we need to look back and molecules and energy
Molecular Motions and Energy n n When a substance is heated, the motion of its molecules increases Molecules can undergo three kinds of motion n The entire molecule can move in one direction n The atoms in the molecule vibrate back and forth n n Called translational motion Molecules in gas have more translational motion than those in their liquid form, which has more than its solid form Called vibrational motion Finally, the molecules may spin n Called rotational motion
Boltzmann Equation n Thermodynamics classically ignored the microscopic structure of matter n n Thermodynamics was a well-developed field before the modern view of atomic and molecular structure was even known In order to link the microscopic and macroscopic descriptions of matter, we use a field of statistical thermodynamics n n Uses statistics and probability to show the link between the two descriptions of matter Will link entropy (a property of bulk matter) to the behavior of atoms and molecules
Mathematical Analysis n n Not going to use math to do this The math gets VERY complex Lucky you Squirrel cannon
Conceptual Analysis n Consider one mole of an ideal gas in a particular state n Defined by specifying T, and V of the gas n n By fixing T and V and n, we also fix(define) P We want to look at this gas sample on the microscopic level n We know from earlier in the year that the particles are constantly in motion n n kinetic-molecular theory The speeds of the gas molecules have well-known speed distributions
Microstate (like Rhode Island) n Now imagine we can take a snapshot of the positions and speeds of all of the molecules at a given instant n n That particular set of 6 x 1023 positions and energies of each gas molecule is called a microstate of thermodynamic system A microstate is a single possible arrangement of the positions and energies of the gas molecules n When we have defined n, V and T
Continue to imagine. . . n Keep taking snapshots of the system, and we would see other microstates n n We use the tools of statistics and probability to determine the total number of microstates for a given thermodynamic state Each thermodynamic state has a number of microstates it is associated with n Which we symbolize with a W
Link to other material n The connection between the number of microstates of a system, W, and its entropy, S, is expressed by Boltzmann’s equation (never use this on test) n S = k(ln. W) n n n k is Boltzmann’s constant 1. 38 x 10 -23 J/K Thus, we use entropy to measure how many microstates are associated with a particular macroscopic state
Given that equation n n The entropy change associated with any process can be given by ΔS = k(ln. Wfinal) - k(ln. Winitial) or So, any change in the system that leads to more microstates leads to a positive value of ΔS n Entropy increases with the number of microstates of the system
Changing our ideal-gas sample n Suppose we increase the volume of the system n n n Which we will view as the same as allowing the gas to expand isothermally Greater volume = more positions available for each atom Thus, more microstates for the system Hence, entropy increases So as volume increases isothermally, entropy increases
Changing system again n Now we keep the volume the same but increase the temperature n Remember than in increase in temperature increases the average (rms) speed of the molecule n n n Broadening the distribution of speeds of the particles Which gives us again more microstates So increasing temperature will increase the entropy of the system
Real Particles n When we consider real molecules (instead of ideal-gas molecules) we need to consider the different amounts of vibrational and rotational energies n n n Not just the kinetic energies A collection of real molecules has more microstates than the same number of ideal-gas particles So real gases have more entropy than ideal-gas versions Additionally, adding molecules also gives more molecules that can change, increasing microstates So adding molecules to a system increases entropy
Viewing Entropy in Terms of Microstates n There a few ways chemists view an increase in microstates/increase in entropy for a system n n Some say increased entropy represents an increase in randomness or disorder Some say that increasing entropy represents an increase in the spreading out of energy
Making Qualitative Predictions about ΔS n We can usually only make qualitative predictions about ΔS when talking about an increase in temperature, volume or particles within a system
Generally When dealing with chemicals, there are three processes we expect to have an increase in entropy n 1. 2. 3. Gases are being formed from either solids or liquids Liquids or solutions are formed from solids The number of gas molecules increases during a chemical reaction
Example n For each of the following, predict whether ΔS is positive or negative, assuming isothermic condtions n H 2 O(l) H 2 O(g) n n n Ag+(aq) + Cl-(aq) Ag. Cl(s) n n n We are forming a solid, which is confined, and has a smaller volume 4 Fe(s) + 3 O 2(g) 2 Fe 2 O 3(s) n n n + ΔS Rule #1 Forming a solid, losing molecules N 2(g) + O 2(g) 2 NO(g) n n No change 2 moles of gas on both sides, no state change
Example #2 n Which has more entropy? n 1 mol of Na. Cl(s) or 1 mol of HCl(g)? n n n 2 mol of HCl(g) or 1 mol of HCl(g)? n n n HCl. Gases have more motions available than solids 2 mol More molecules means more entropy 1 mol of HCl(g) or 1 mol of Ar(g) n n HCl molecules can rotate and vibrate. Ar atoms cannot. You need molecules to rotate and vibrate.
The Third Law of Thermodynamics n If we decrease thermal energy of a system by lowering the temperature n n n The energy stored in translational, vibrational and rotational forms of motion decrease As less energy is stored, the entropy of the system decreases If we eventually lower the temperature enough to reach a state where these motions effectively stop, we reach a single microstate n Which means zero entropy
n This leads to the third law of thermodynamics n States that the entropy of a pure crystalline substance at absolute zero is zero n n S(0 K) = 0 Remember, S = k (ln. W) n n So when W = 1 S = k(ln 1) = 0
Heating and Entropy n Let’s see what happens now when we heat a substance that was at T = 0 n As we heat the solid, T goes up, so entropy goes up n Until the melting point. Here the bonds holding the atoms or molecules together are broken, and particles are free to move. n n Same thing occurs as we heat the liquid n n No change in T while melting, but S increases, but while boiling, S goes up while T stays the same And same while we heat the gas
Just to reiterate. . . n When comparing the solid, liquid and gaseous form of a substance n Ssolid < Sliquid < Sgas
17. 6 – Entropy Changes in Chemical Reactions n n Earlier in the year we discussed how calorimetry can be used to measure ΔH for a chemical reaction. Unfortunately, there is no comparable way to measure ΔS n But by using experimental measurements and looking at the variation in heat capacity with temperature, we can find the absolute vale of the entropy, S
Absolute Entropy n Absolute entropies are based on the reference point of 0 entropy at 0 K n n Third law of thermodynamics Entropies will usually be given as molar quantities n n Which we call molar entropies Units of J/mol-K
Molar Entropies n Molar entropy values of substances in their standard states are known as standard molar entropies n n Given the variable Sº Remember, the standard state for a substance is defined as the pure substance at 1 atm pressure and usually 298 K n Appendix IIB has entropy values
Generalizations about Molar Entropies 1. 2. 3. 4. Unlike enthalpies of formation, the standard molar entropies of elements is NOT zero Standard molar entropies of gases are greater than liquids and solids They generally increase with increasing molar mass Generally increase with increasing number of atoms in the formula of the substance
Entropy Change n n The entropy change in a chemical reaction equals the sum of the entropies of the products minus the sum of the entropies of the reactants ΔSº=Σn. Sº(products) - Σ m. Sº(reactants) n n and m are the coefficients in the chemical equation
Example n Calculate ΔSº for the synthesis of ammonia from N 2(g) and H 2(g) at 298 K n n N 2(g) + 3 H 2(g) 2 NH 3(g) ΔSºNitrogen = 191. 5 ΔSºHydrogen = 130. 6 ΔSºAmmonia = 192. 5
n n n Use the equation ΔSº=Σn. Sº(products) - Σ m. Sº(reactants) ΔSº= 2 Sº(NH 3) - [Sº(N 2) + 3 Sº(H 2)] ΔSº= 2(192. 5) - [1(191. 5) + 3(130. 6) ΔSº= -198. 3 J/K n n This should make sense, since we went from 4 molecules of reactant to 2 molecules of product Decrease in number of molecules = decrease in entropy
17. 5 - Gibbs Free Energy** n n n So far, we have seen spontaneous, endothermic reactions must increase the entropy of the system Additionally, we have seen spontaneous, exothermic reactions that can actually cause a decrease in entropy of the system Therefore, spontaneity involves two thermodynamic concepts n Entropy and enthalpy (exothermic or endothermic)
n n We can use ΔH and ΔS to predict whether a given reaction occurring at a given temperature and pressure will be spontaneous. The way to do this was developed by an American mathematician named Willard Gibbs n He proposed a new state function, now called the Gibbs free energy (or simply just free energy).
Gibbs Free Energy n The free energy (G) of a state is n n n G = H - TS T is absolute temperature At a constant temperature, the Gibbs free energy of a system, ΔG is n ΔG = ΔH - TΔS
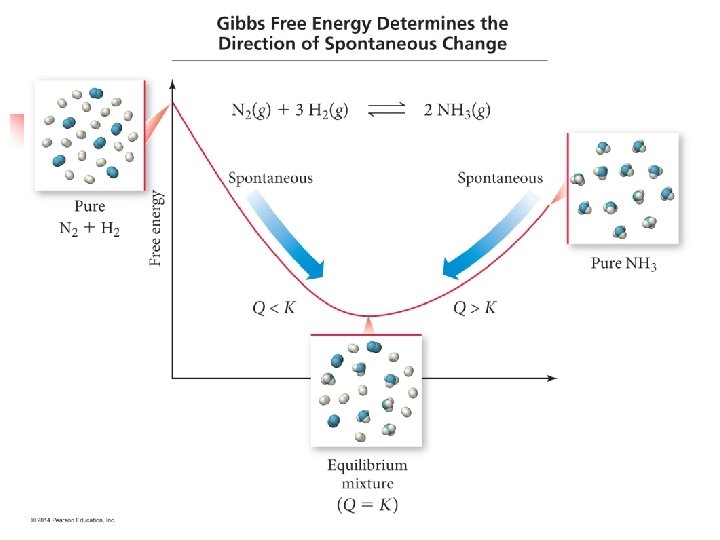
Sign of ΔG n 1. 2. 3. Assuming that both T and P are constant, we can define the relationship between ΔG and process spontaneity If ΔG is negative, the reaction is spontaneous in the forward direction If ΔG is zero, the reaction is at equilibrium If ΔG is positive, the reaction in the forward direction is nonspontaneous; work must be supplied from surroundings to make it occur. Reverse reaction will be spontaneous
Why use this? n It is generally more convenient to use ΔG as the judge for spontaneity than to use Suniverse n Because ΔG relates to the system alone, and avoids having to deal with the surroundings
Analogy n n Often an analogy is used between the freeenergy change during a spontaneous reaction and the potential-energy change when a boulder rolls down a hill Rule: In any spontaneous process at constant temperature and pressure, the free energy always decreases
Haber Process Example n n N 2(g) + 3 H 2(g) ⇄ 2 NH 3(g) Imagine we can maintain constant P and T n What happens when we add a certain number of moles of N 2 and 3 x as many moles of H 2? n n What happens when we fill the container with pure NH 3? n n They will react spontaneously to form NH 3 until equilibrium is reached Will decompose into N 2 and H 2 until equilibrium In both cases, the free energy of the system is lowered on the way to equilibrium
17. 7 - Standard Free-Energy Changes in Chemical Reactions n Since free energy is a state function (like enthalpy) we can tabulate standard free energies of formation for substances n Remember that these standard values imply certain conditions, or standard states n n n For gaseous substances, 1 atm pressure For solid substances, we assume a pure solid For liquid, a pure liquid For solutions, generally 1 M Generally, temperature will be 25ºC, but not always For elements, free energies of elements in standard state is zero (just like with enthalpy)
Can We Really be That Arbitrary? n n Choosing a standard state really is an arbitrary decision But it is okay n n n Because the standard state is a reference point And we are primarily dealing with the difference in free energies between reactants and products A list of free energies of formation, ΔGºf is found in Appendix II-B
Calculating standard free energies n n Again, we usually use the free energy change ΔGº = ΣnΔGºf(products) - ΣnΔGºf(reactants) n n And again, n and m are the coefficients in the equation Really, very similar to the ΔHº calculations we have done already
Example n Calculate the free energy change for the following reaction at 298 K and determine if this reaction is spontaneous n n P 4(g) + 6 Cl 2(g) 4 PCl 3(g) From appendix II n n n ΔGºf of PCl 3(g) = -269. 6 k. J/mol ΔGºf of P 4(g) = 24. 4 k. J/mol ΔGºf of Cl 2(g) = 0 k. J/mol
n n n ΔGº = ΣnΔGºf(products) - ΣnΔGºf(reactants) = 4 ΔGºf[PCl 3(g)] - ΔGºf[P 4(g)] - 6 ΔGºf[Cl 2(g)] = 4(-269. 6 k. J/mol) - 1(24. 4 k. J/mol) - 0 =-1102. 8 k. J So this reaction would be spontaneous
Example 2 n n n Estimate whether ΔGº for the following reaction is more or less negative than ΔHº. C 3 H 8(g) + 5 O 2(g) 3 CO 2(g) + 4 H 2 O(l) ΔHº = -2220 k. J n Free energy incorporates both the change in enthalpy and the change in entropy for a reaction n n ΔGº = ΔHº - TΔSº We will need to determine the sign of TΔSº n We do this by looking at the reaction
ΔG = ΔH - TΔS n C 3 H 8(g) + 5 O 2(g) 3 CO 2(g) + 4 H 2 O(l) n n Reactants have 6 molecules of gas Products have 3 of gas and 4 of liquid n n n # of gas molecules down significantly This would lead us to believe a significant decrease in entropy of system Therefore, the ΔSº in TΔSº would be negative Because we are subtracting TΔSº, which is negative, we are adding. This means that ΔGº will be less negative than ΔHº
n In appendix II, we can calculate ΔGº for reactions at 25ºC n n n But sometimes we want reactions at other temperatures We can use either free energy equation to see what happens at other temperatures ΔG = ΔH TΔS enthalpy entropy term
ΔG = ΔH - TΔS n n ΔG will vary with temperature T is positive number for all temperatures n n n Except for abolsute zero ΔH can be positive or negative TΔS can also be positive or negative n When ΔS is positive, we get more entropy than in the initial state, and -TΔS will be negative
The Rules: Pt. 1 n The sign of ΔG tells us if a process is spontaneous n n n Depends on the magnitude of ΔH and -TΔS When both ΔH and -TΔS are negative, ΔG will be negative and the process will be spontaneous at ALL temperatures When ΔH and -TΔS are positive, ΔG will be positive and the process will be nonspontaneous at all temperatures
The Rules: Continued n When ΔH and -TΔS have opposite signs, the sign of ΔG will depend on the magnitude of the two terms n n Here is where temperature becomes important Generally, ΔH and ΔS change little with temperature n n But value of T directly affects the magnitude of -TΔS As T increases, magnitude of -TΔS increases and will become relatively more important when finding the sign and magnitude of ΔG
n n Consider the following process H 2 O(s) H 2 O(l) ΔH > 0, ΔS > 0 n n n Process is endothermic, which means ΔH is positive Entropy increases, so ΔS is also positive and therefore TΔS is negative At temperatures below 273 K (0ºC), the magnitude of ΔH is greater than that of -TΔS n n So enthalpy term dominates, and ΔG remains positive This means that the melting of ice is nonspontaneous at temperatures below 0ºC, which we expect
However n What happens in this process when the temperature is greater than 0ºC? n n The magnitude of the entropy term -TΔS increases As the entropy term increases, it will become greater than the magnitude of ΔH. n n So now the entropy term dominates, which gives us a negative value for ΔG. So at temperatures above 0ºC, we see that the melting of ice is spontaneous.
The Rules n ΔH Effects of temperature on spontaneity is summarized by the following table ΔS -TΔS ΔG Reaction Characteristics - + - - Spontaneous at all temperatures + - + + Nonspontaneous at all temperatures - - + +/- Spontaneous at low T; nonspontaneous at high T + + - +/- Spontaneous at high T; nonspontaneous at low T
Example n Haber process n n N 2(g) + 3 H 2(g) ⇄ 2 NH 3(g) Assume that ΔHº and ΔSº do not change with temperature. What is the direction in which ΔGº for this reaction changes with increasing temperature. What is the value of ΔGº for this reaction at 25ºC and 500ºC n Note: ΔHº = -92. 38 k. J and ΔSº = -198. 4 J/K
Direction on free energy change with increasing temperature n Both ΔHº and ΔSº are negative for this process n n Because ΔSº is negative, the -TΔS is positive. So we get increasing ΔGº values with increasing T
Calculation of ΔGº n n At T = 25ºC = 298 K ΔGº = ΔHº -TΔSº n n = -92. 38 k. J - (298 K)(-198. 4 J/K)(1 k. J/1000 J) = -92. 38 k. J + 59. 1 k. J = -33. 3 k. J At T = 500ºC = 773 K ΔGº = ΔHº -TΔSº n n n = -92. 38 k. J - (773 K)(-198. 4 J/K)(1 k. J/1000 J) = -92. 38 k. J + 153 k. J = 61 k. J This tells us that at some point between these two temperatures, this reaction goes from being spontaneous to nonspontaneous
17. 8 - Free Energy and the Equilibrium Constant*** n Earlier we saw a relationship between ΔG and equilibrium n n n For a system at equilibrium, ΔG = 0 We can also use thermodynamic data (like in appendix II) to find values of the standard free-energy change, ΔGº For this section, we will see how to use free energy to find the value of ΔGº under nonstandard conditions and to relate ΔGº to the equilibrium constant for the reaction
Nonstandard Conditions n The set of standard conditions for which we can find ΔGº is in appendix II n n n However, most chemical reactions occur under nonstandard conditions. For any chemical process, the general relationship between ΔGº and the free energy change of any other conditions ΔG is found by ΔG = ΔGº + RT(ln. Q) n n n R is ideal gas constant, 8. 314 J/mol-K T is absolute temperature Q is the reaction quotient that goes along with the particular reaction mixture n Remember that finding Q is just like finding the equilibrium constant, except that we use initial concentrations, not equilibrium concentrations
ΔG = ΔGº + RT(ln. Q) n Under standard conditions, the concentrations of all reactants and products are equal to 1 M n n So under the standard conditions Q = 1 and therefore ln. Q = 0 Which means that ΔG = ΔGº under standard conditions
Example n What is the value of ΔGº for the following equilibrium? n CCl 4(l) ⇄ CCl 4(g) n n n At equilibrium, ΔG = 0 Since we are in equilibrium, Q = 1 And then ln. Q = 0 So ΔG = ΔGº Therefore, ΔGº = 0
n When the concentrations of reactants and products are nonstandard, we must calculate Q to find ΔG.
Example n Using the Haber process, what is ΔG at 298 K for a reaction that consists of 1. 0 atm N 2, 3. 0 atm H 2 and 0. 50 atm NH 3? n n N 2(g) + 3 H 2(g) ⇄ 2 NH 3(g) ΔGº = -33. 3 k. J/mol
n Find Q n Now plug into equation ΔG = ΔGº + RT(ln. Q) n ΔG = (-33. 3 k. J/mol) + (8. 314 J/mol-K)(298)(1 k. J/1000 J)(ln 9. 3 x 10 -3) n ΔGº = (-33. 3 k. J/mol) + (-11. 6 k. J/mol) = -44. 9 k. J/mol n
Bringing in Equilibrium n We can now use our previous equation to find the relationship between ΔGº and the equilibrium constant, K n n At equilibrium, ΔG = 0 Equilibrium quotient, Q, equals equilibrium constant, K, when system is at equilibrium
At Equilibrium n At equilibrium, we transform our equation n Therefore, n n ΔG = ΔGº + RT(ln. Q) into 0 = ΔGº + RT(ln. K) ΔGº = -RT(ln. K) And if we know ΔGº, we can solve for K n K = e-ΔGº/RT
Note about these equations n We have to be careful with our units n n ΔGº should be in k. J/mol Gas pressures should be in atm Concentration measured in molarity Solids, liquids and solvents do not appear in our equation n Like with normal equilibrium problems
ΔGº = -RT(ln. K) n We can see that if ΔGº is negative n n n Then ln. K must be positive A positive value for ln K means that K > 1 Therefore, the more negative ΔGº is, the larger the equilibrium constant Which means the more the reaction favors the products Also, if ΔGº is positive n n Then ln. K is negative, so K < 1 Therefore we favor the reactants
Example n Use standard free energies of formation to calculate the equilibrium constant, K at 25ºC for the Haber process n n N 2(g) + 3 H 2(g) ⇄ 2 NH 3(g) ΔGº = -33. 3 k. J/mol
n K = e-ΔGº/RT n n Solving for the exponent -ΔGº/RT -(33, 300 J/mol)/(8. 314 J/mol-K)(298 K) = 13. 4 K = e 13. 4 = 7 x 105