Differential diagnosis and treatment strategy of neuromuscular disorders


 • AIDP • AMAN • AMSAN • Acute sensory neuronopathy")
 • Classical CIDP, axonal CIDP, sensory CIDP •")
 neuropathies •")
")
 •")









 • Multifocal acquired demyelinating sensory and motor (MADSAM) neuropathy • Predominant")
 • • • Oculomotor nerve palsies for 2 patients (26%) Multifocal")
 • A very focal motor fiber demyelination sparing the nerve")









 vs CIDP (46) POEMS CIDP Polyneuropathy Symmetric Demyelination 70% definite CIDP")

 Ig. M MGUS (60%): • Bone marrow biopsy")






 15 -20%")

, CMTX (GJB 1), HNPP (PMP")
















 - Pre-synatic disorder LEMS - Subacute onset, - Fluctuation less")


- Slides: 69
Differential diagnosis and treatment strategy of neuromuscular disorders 2017住院醫師教育課程 中醫大附醫 郭育呈 20170820
Outline • Classification and clinical manifestations of dysimmune neuropathies • Diagnosis of dysimmune neuropathies • Treatment of dysimmune neuropathies • Autoimmune myasthenia gravis • ACh. R Ab, Mu. SK Ab, LRP 4 Ab • Lambert-Eaton myasthenic syndrome (LEMS) 2
Classification-I Guillain-Barre syndrome (GBS) • AIDP • AMAN • AMSAN • Acute sensory neuronopathy • Acute pan-dysautonomia • Regional variants: MFS, oropharyngeal • Overlap: Miller Fisher-Guillain Barre overlap syndrome • Acta Neurol Scand 2005; 112: 115 -125. • Nat Clin Pract Neurol 2007; 3: 198 -211. 3
Classification-II Chronic inflammatory demyelinating polyneuropathy (CIDP) • Classical CIDP, axonal CIDP, sensory CIDP • CIDP with DM • CIDP with MGUS (Ig. M, Ig. G, Ig. A) • MMN with and without conduction block • MADSAM (Lewis-Sumner syndrome) • MASAM • Distal acquired demyelinating sensory (DADS) neuropathy • Acta Neurol Scand 2005; 112: 115 -125. 4
Classification-III Paraprotein-associated neuropathy, monoclonal gammopathies • Waldenstrom’s macroglobulinemia • Anti-myelin-associated glycoprotein (MAG) neuropathies • Polyneuropathy, organomegaly, endocrinopathy, M -protein, skin changes (POEMS) syndrome • Mixed cryoglobulinemia • Gait ataxia, late-onset polyneuropathy (GALOP) syndrome • Monoclonal gammopathy with undetermined significance (MGUS) • Acta Neurol Scand 2005; 112: 115 -125. 5
Diagnostic methods Clinical phenotypes Electrophysiological findings CSF study- protein, Ig. G index (inherited, vasculitis) Serum protein electrophoresis (S-PEP)- serum immunoelectrophoresis (S-IEP) & serum immunofixation electrophoresis (S-IFE) • Antibodies in the serum • Pathology • • 6
Autoantibodies in the serum • Monoclonal antibodies without identifiable antigenic targets (M proteins) • Monoclonal and polyclonal autoantibodies that bind to specific neural components • Diagnostic utility • Provide clues to the pathogenesis of neuropathic syndrome • Suggest specific avenues of therapy 7
Guillain Barre syndrome • GBS always follows an infection as URI or GI infection • Campylobacter jejuni, CMV, EBV, mycoplasma pneumonia • Vaccination and stress events debated • 25% artificial ventilation (vital capacity < 15 -20 ml/kg) • 2/3 ANS dysfunction • Urinary retention and constipation are unusual at the onset of the disease but commonly develop at the nadir of the disease. • Autoimmunity Reviews 2014; 13: 525 -530. 8
- 2/3 - 80% > 1 week - GBS at nadir Asbury et al, Ann Neurol 1990
Lancet Neurol 2008
Antibodies to gangliosides Lancet Neurol 2008
Guillain Barre syndrome • 20% disability after 1 year • Adverse prognostic factors: advanced age, the severity of disease at nadir, bed-bound, artificial ventilation. • Plasma exchange or IVIg (0. 4 gm/kg/day in 5 days) • Oral steroid or pulse therapy with methylprednisolone alone not beneficial • Autoimmunity Reviews 2014; 13: 525 -530. 12
Multifocal motor neuropathy 1 -2/100, 000, male > female, mean onset 40 y/o Pure motor, asymmetric weakness 80% initially affected forearm/hands, 10% LLs 50% fasciculation and cramp, prominent than MND Hypo-reflexia (patchy or diffuse), 20 -30% normo/hyper-reflexia • Minor vibration impairment of lower limbs • • • Brain 2002; 125: 2591 -2625. 13
Multifocal motor neuropathy • • • Multifocal conduction blocks 30 -80% anti-GM 1 Ig. M antibody (not specific) MMN without conduction block-> IVIg good response IVIg is the first-line therapy Uncontrolled studies: cyclophosphamide, interferon β-1 a, cyclosporine, methotrexate, azathioprine and Rituximab • Cochrane review • Autoimmun Rev 2014; 13: 525 -530. 14
EFNS criteria -JPNS 2010; 15: 295 -301. Autoimmun Rev 2014; 13: 525 -530 15
16
MADSAM (Lewis-Sumner syndrome) • Multifocal acquired demyelinating sensory and motor (MADSAM) neuropathy • Predominant distal, asymmetric weakness most affecting upper limbs with sensory impairment • Initial onset: distal part of an upper limb (70%) • Numbness confined to dermatome (not stocking-glove) • Mononeuropathy multiplex, demyelinating S-M poly • Hypo-reflexia with multifocal & asymmetric pattern, minority of patients with generalized hypo-reflexia • MN 1999; 22: 560 -566. MN 2005; 31: 88 -94. • Brain 2004; 127: 2010 -2017. 17
MADSAM (Lewis-Sumner syndrome) • • • Oculomotor nerve palsies for 2 patients (26%) Multifocal conduction block and demyelination (forearm) 87% abnormal distal sensory potentials (sural n SNAPs) 82% elevated CSF protein (MMN 9%) No anti-GM 1 antibody (MMN 56%) Sural nerve biopsy: prominent demyelination (MMN no) • MN 1999; 22: 560 -566. • Brain 2004; 127: 2010 -2017. 18
MADSAM (Lewis Sumner syndrome) • A very focal motor fiber demyelination sparing the nerve endings, whereas sensory fiber involvement was widespread. • Course: chronic progressive in 71% , relapsing-remitting in the others. • IVIg 55%, 33% oral steroids 73% positive response to immune-mediated therapy • Brain 2004; 127: 2010 -2017. 19
• MN 1999; 22: 560 -566. 20
CIDP DADS neuropathy MADSAM neuropathy MMN Weakness Symmetric, P+D Symmetric, D, Mild or no weakness Asymmetric, D > P, Upper > Lower Sensory Symmetric Multifocal (nerve) No DTR Reduced, symmetric Reduced (multifocal or diffuse) NCV: demyelinating Symmetric, Prolonged DL Asymmetric (multifocal) CB Frequent (-) Frequent Abnormal SNAPs Symmetric Uncommon Asymmetric (multifocal) SNAPs normal CSF proteins High Normal Monoclonal protein Ig. G or Ig. A Ig. M-κ, 50 -70% MAG (+) Rare Anti-GM 1 Ab Rare (-) Rare 50% (+), Ig. M Prednisolone Yes Poor Yes No PLEX Yes Poor Possible No IVIg Yes Poor Yes Cyclophosphamide Yes Poor Possible Yes • Neurol Clin 2013; 31: 533 -555. 21
Paraproteinemic demyelinating polyneuropathy • Clinical phenotypes • Immunoglobulin class • Monoclonal gammopathy of undertermined significance or malignant plasma cell dyscrasia • Presence of antibodies to MAG • Electrophysiology • Likelihood that paraprotein is causing the neuropathy • EFNS 2006 guideline. Euro J Neurol 2006; 13: 809 -818. 22
Paraproteinemic demyelinating polyneuropathy • Paraprotein: an underlying disease of plasma cells in bone marrow (malignant or MGUS) • Ig. M paraproteinemia vs Ig. G/Ig. A • Heavy chain (Ig. M, Ig. G, or Ig. A) and light chain (kappa or lambda) • EFNS 2006 guideline. Euro J Neurol 2006; 13: 809 -818. 23
Classification of hematological conditions with a paraprotein Malignant monoclonal gammopathies • Multiple myeloma (overt, asymptomatic (smouldering), nonsecretory, or osteo-slcerotic) • Plasmacytoma (solitary, extramedullary, multiple solitary) • Malignant lympho-proliferative disease: - Waldenstrom’s macroglobulinemia - Malignant lymphoma - Chronic lymphocytic leukemia • Heavy chain disease • Primary amyloidosis (AL) (with or without myeloma) Monoclonal gammopathy of undertermined significance (MGUS) • EFNS 2006 guideline. Euro J Neurol 2006; 13: 809 -818. 24
Investigation of paraprotein • Serum IFE • CBC-DC, BUN/Cr, AST/ALT, Ca, P, UA, ESR, CRP, LDH, RF, β 2 -microglobulin, RF and cryoglobulins • Immunoglobulin Ig. G, Ig. A and Ig. M • Urine for Bence-Jones protein (free light chains), and, if positive 24 hour urine for protein quantification • Skeletal X ray: skull, pelvis, spine, ribs & long bones (shoulder to wrist & hip to ankle), 99 m. Tc MIBI scan. • Sonography or CT scan of abdomen & chest • Bone marrow aspiration • EFNS 2006 guideline. Euro J Neurol 2006; 13: 809 -818. 25
Investigation of paraprotein- PE • • Lymphadenopathy Hepato-splenomegaly Macroglossia Signs of POEMS syndrome • EFNS 2006 guideline. Eur J Neurol 2006; 13: 809 -818. 26
POEMS syndrome Lytic lesions osteosclerotic lesions • Clinical lymphoma myeloma leukemia 2015; 15: e 15 -21. 27
POEMS syndrome • Demyelinating polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes • Plasma cell dyscrasia and multiple organ disorder • Overproduction of VEGF by plasmacytomas • 50% initial presentation with polyneuropathy, many are misdiagnosed as CIDP. • CSF: albuminocytological dissociation • JNNP 2012; 83: 476 -479. 28
POEMS syndrome (51) vs CIDP (46) POEMS CIDP Polyneuropathy Symmetric Demyelination 70% definite CIDP criteria Albuminocytological dissociation; paraprotein 70%; λ M-protein Neuropathy onset 49%, 60% initial CIDP Dx Severe leg pain 76% 7% Muscle atrophy 52% 24% Distal weakness 52% 24% Median nerve- prolonged DL less Median nerve- TLI higher Tibial and sural nerves No response Conduction block/temporal dispersion Rare (6%) • JNNP 2012; 83: 476 -479. JNNP 2012; 83; 480 -486. • MN 2002; 26: 189 -193. 30
POEMS syndrome vs CIDP POEMS CIDP Polyneuropathy Symmetric Demyelination 70% definite CIDP criteria Albuminocytological dissociation 70% Neuropathy onset 49%, 60% initial CIDP Dx • Demyelination predominant in the nerve 7% trunk rather than 76% in the distal nerve terminals. Muscle atrophy 52% 24% • Axonal loss in the lower limb nerves. Severe leg pain Distal weakness 52% 24% Median nerve- prolonged DL less Median nerve- TLI Higher ? recheck Tibial and sural nerves No response • JNNP 2012; 83: 476 -479. 31
Monoclonal gammopathy of undetermined significance (MGUS) Ig. M MGUS (60%): • Bone marrow biopsy normal • No S/S of tumor infiltration (constitutional symptoms, hyper-viscosity, organomegaly) • Observation 12 months Ig. G or Ig. A MGUS (Ig. G 30%, Ig. A 10%): • Monoclonal component ≤ 30 g/L (3 g/d. L) • Bence-Jones proteinuria ≤ 1 g/24 h • No lytic lesions in bone; bone marrow plasma cell <10% • No anemia, hyper-Ca or chronic renal insufficiency • Observation 12 months • EFNS 2006 guideline. Eur J Neurol 2006; 13: 809 -818. 32
Nat Neurol Rev 2014
Treatment • • • GBS: IVIg, plasmapheresis CIDP: IVIg, steroid, plasmapheresis MMN: IVIg MADSAM: IVIg, steroid POEMS: steroid, add-on IS, plasmapheresis MGUS: steroid, plasmapheresis, 2 nd imuran 34
Hereditary or acquired ? Hereditary Pattern diffuse, homogenous Acquired disproportionate, asymmetric Temporal dispersion limited excessive Demyelination uniform segmental Family history AD, AR, sporadic
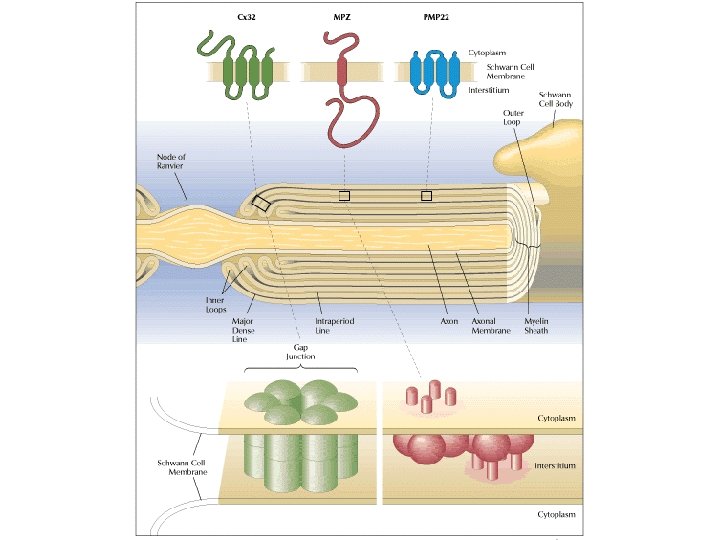
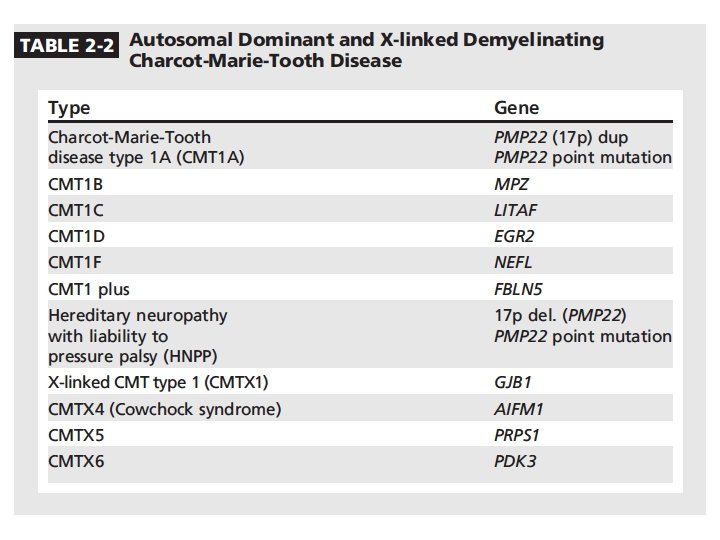
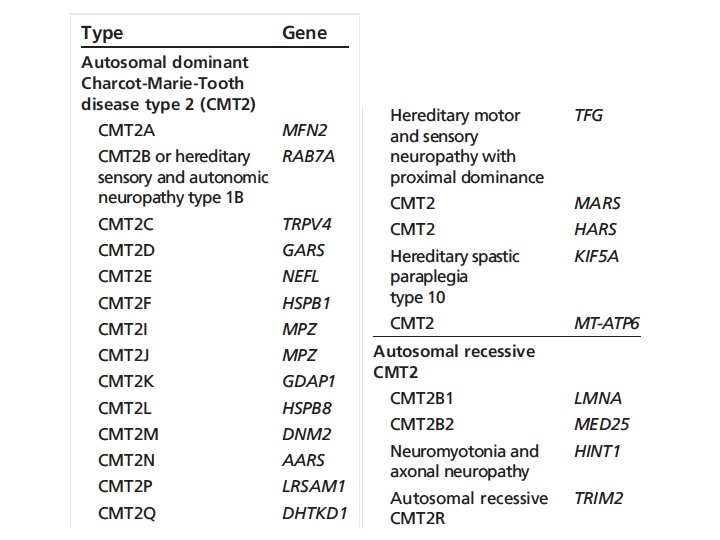
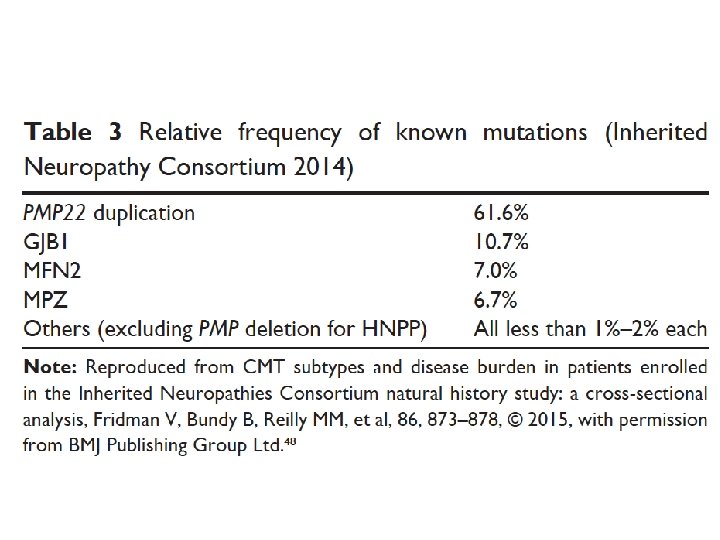
Charcot-Marie-Tooth diseases • Mutations in the same gene can manifest as distinct phenotypes, the same phenotype can be caused by mutation in different genes • Incidence 1/2500 in western countries • Insidious onset and progression • AD, X-linked, AR • 90% of CMT: PMP 22, GJB 1, MFN 2, MPZ • Demyelinating CMT 1: 50% cases • Norway study: CMT 1 (37%), CMT 2 (35. 9%)
Median nerve conduction velocities • CMT 1: AD demyelinating CMT 2: AD axonal CMTX: X-linked CMT 4: AR CMT • CMT 3: early-onset, Dejerine-Sottas disease • < 38 m/s CMT 1 demyelinating • > 38 m/s CMT 2 axonal • 35 -45 m/s intermediate conduction velocities (GJB 1, DNM 2, YARS, MPZ, IFN 2, GNB 4)
CMT 1 A • CMT 1 A: Absent sensory responses, reduced motor conduction velocities (~25 m/s), reduced amplitudes (secondary axonal loss) • PMP 22 duplication • Rare require a wheel-chair during the lifetime
CMT-X 1 • CMTX 1, GJB 1, gap junction protein (connexin 32) 15 -20% of CMT. • CMTX 1: male more severe, female mild pattern • CMTX 1: split hand syndrome (APB wasting >> FDI) • Asymmetric pattern: HNPP, Acquired inflammatory neuropathies, GJB 1 • CMTX 1: intermediate nerve conduction velocities • Acute CNS dysfunction as stroke-like event with MRI changes
CMT 2 • 25 -30% of CMT • CMT 2 A, MFN 2 gene, 20% of axonal CMT, most common type • CMT 2 A: 23/27 cases non-ambulatory before 20 year old
• CMT: CMT 1 A (PMP 22 duplication), CMTX (GJB 1), HNPP (PMP 22 deletion), MFN 2 • CMT 1: CMT 1 A (PMP 22 duplication), HNPP (PMP 22 deletion), CMT 1 B (MPZ) • HNPP: marked slowing of ulnar & sural sensory NCV with or without reduced SNAP amplitudes, relatively preserved motor NCV, prolonged DLs in median and peroneal nerves crossing knees, CBs and focal slowing at entrapment sites • CMT 1 X: asymmetric pattern, no episodic attacks
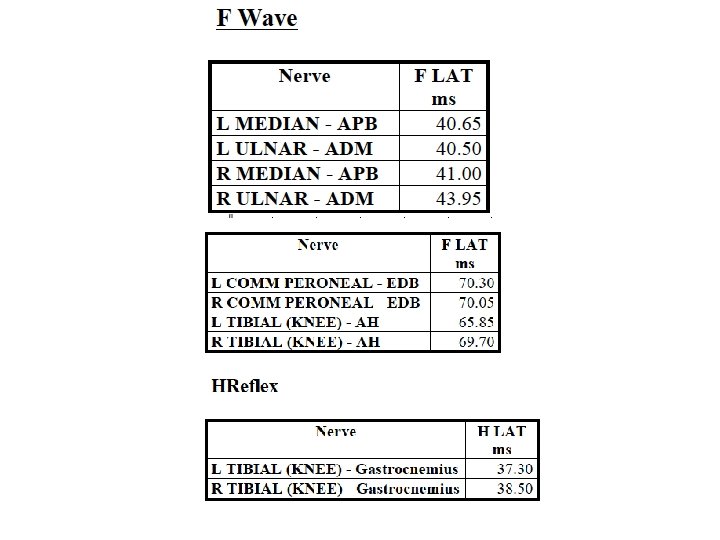
# H-reflex: no responses, bilateral.
Microsattelite Result: PMP 22 duplication.
# 16 y/o boy, Right upper limb weakness after 拔河.
HNPP
Familial amyloid neuropathy
# Hot spot mutation in Taiwan: Ala 97 Ser JNNP 2015
JPNS 2016
Misdiagnosis of FAP for CIDP • Treatment-unresponsiveness • Severe autonomic dysfunction • Weight loss • Hx of carpal tunnel syndrome Red flags
Red flags for TTR-FAP
Myasthenia gravis • Autoantibodies to post-synaptic muscle endplates; B-cell mediated • Acetylcholine receptor (ACh. R), muscle-specific kinase (Mu. SK), lipoprotein-related protein 4 (LRP 4) antibodies • Changes in antibody concentration might predict disease severity. • 10 -15% of patients- full control is not possible • Lancet Neurology 2015; 14: 1023 -1036. 61
Lancet Neurol 2015
• • Lancet Neurol 2015 60% ptosis or diplopia, or both. 20% restricted to ocular MG 90% of ocular MG remain in ocular type for > 2 years EOM-asymmetric, limbs weakness-symmetric
MG classification ACh. R Mu. SK LRP 4 Percentage 80% 4% 2% Population (50 y/o) Early onset: F>M (3/1) Late onset: M>F Young females Weakness Ocular or generalized Bulbar and facial (40%) Ocular and limbs: less common Respiratory crisis Ocular (20%) or generalized Mild-to-moderate Thymus pathology Early onset: hyperplasia Late onset: normal No No Thymectomy response Early onset: good Late onset: poor Correlation of Ab titer to severity No Yes ? # Thymoma-associated MG: 10 -15% of all MG, ACh. R Ab(+), generalized type. 30% of patients with thymoma will develop MG. NEJM 2017
Lancet Neurol 2015
Lancet Neurol 2015
Lambert-Eaton myasthenic syndrome (LEMS) - Pre-synatic disorder LEMS - Subacute onset, - Fluctuation less prominent - Favored LEMS instead of MG - Late manifestation vs at onset (MG) 3 A’s: Areflexia, Autonomic dysfunction, Apraxia (functional difficulties of gait, with mild proximal leg weakness) - NCV/EMG Triad -less than >50% of lower limits -Later than 4 th/5 th stimulation (MG) -10% negative J Neurol 2017
MVC: maximal voluntary contraction, 10 s. Ulnar MNCV, ADM Continuum 2016
Thank you for your attention ! 70