LEUKEMIA CHRONIC MYELOID LEUKEMIA Dr Kiran H S






 • 15% of all leukemia • is an acquired clonal")


 • Its an acquired chromosomal abnormality present")
 • t(9; 22)(q 34; q 11) in")
 Altered adhesion Mitogenic activation Inhibition of apoptosis • No adhesion")


 Clinical features 17")








 – Detect the BCR-ABL fusion")
 – Most sensitive test to identify")





 – Intravascular flow-impeding effects of white cell")







 Death is usually")
- Slides: 44
LEUKEMIA- CHRONIC MYELOID LEUKEMIA Dr. Kiran H S Assistant Professor Pathology, YMC
OVERVIEW • • • Epidemiology Etiology & molecular pathogenesis Clinical features Diagnosis Course of disease Treatment
Epidemiology • Commonest leukemia constituting about 2530% of all Leukemias in India. • 5 th to 6 th decade. • 50 -70% of leukemias in India • Male predominance (1. 4: 1) • About 50%- asymptomatic 4
Chronic Lymphoid Leukemias & Myeloproliferative Disorders
Chronic Myeloproliferative Disorders • are malignant clonal proliferations of multipotent stem cells. While all three cell lines (myeloid, erythroid and megakaryocytic) are involved in each disorder, each has specific genetic abnormalities. • As a result the dominant cell differs in each allowing for the subclassification of the chronic myeloproliferative disorders. • Chronic myeloid leukemia (CML), • Polycythemia vera (PV), • Primary myelofibrosis (MF) and • Essential thrombocythemia (ET)
Chronic myeloproliferative disorders are • acquired, malignant clonal disorders • characterized by expansion of pluripotent stem cells showing deranged production of one or more myeloid lines and having a variable predisposition to transform to leukemia.
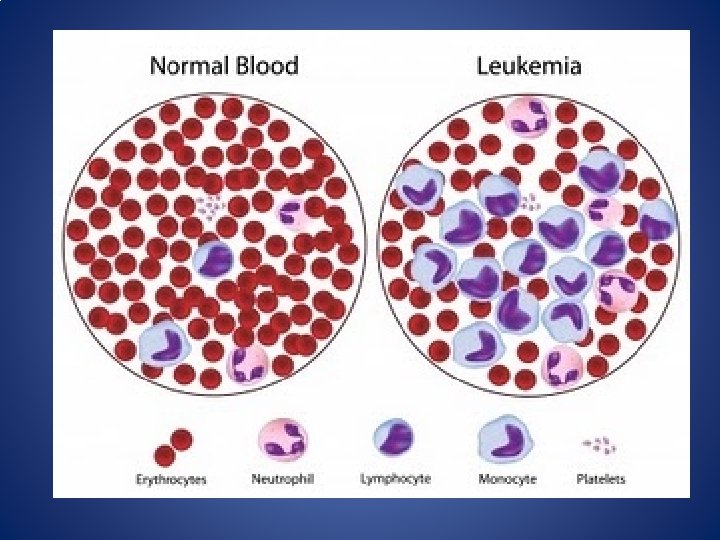
Chronic Myeloid Leukemia (CML) • 15% of all leukemia • is an acquired clonal MPN of the abnormal pluripotent hematopoietic stem cell. • Malignant disorder of multipotent stem cells with predominance of mature granulocytes and their precursors accumulating in excess in the marrow and blood.
• It is distinguished by the presence of chimeric fusion BCR-ABL gene & characteristic chromosomal abnormality i. e Philadelphia(Ph) chromosome in more than 90% of cases.
Etiology and Pathogenesis • Not known. • Exposure to ionising radiation may increase the risk and is dose dependent.
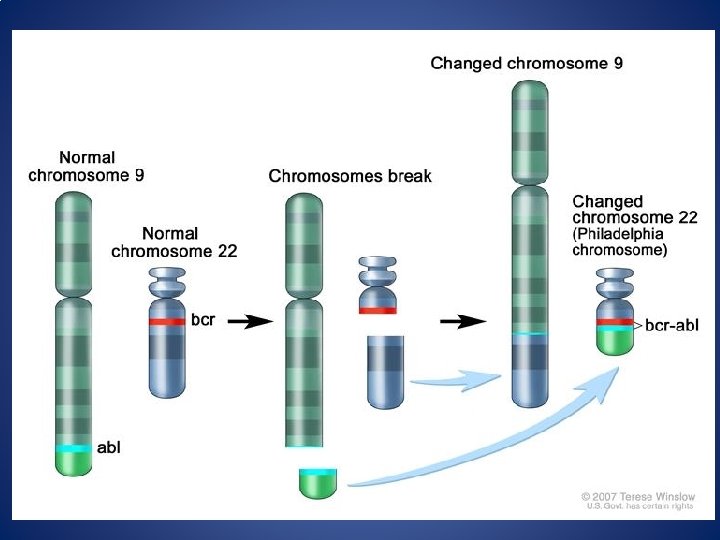
MOLECULAR PATHOGENESIS: CML & Philadelphia chromosome (Ph') • Its an acquired chromosomal abnormality present in all proliferating hematopoietic stem cells. • Balanced reciprocal translocation between chromosome 9 and 22… t(9, 22)(q 34; q 11. 2) • Translocation results in increase in length of chromosome 9 and shortened long arm of one of the chromosome 22. • Shortened chromosome 22 – Ph chromosome.
MOLECULAR PATHOGENESIS: CML & Philadelphia chromosome (Ph') • t(9; 22)(q 34; q 11) in which the c-abl gene from 9 is juxtaposed with the bcr sequence on 22. • This leads to a chimeric gene (bcr-abl) the product of which is an aberrant 210 k. D tyrosine kinase (thought to be involved in cell regulation). • Mature myeloid cells appear to live longer and thus accumulate. bcr-abl gene product
3. BCR-ABL (Tyrosine Kinase) Altered adhesion Mitogenic activation Inhibition of apoptosis • No adhesion to marrow stroma • Reduced regulation by marrow factors • Activation of various pathways proliferation • Upregulation of Bcl-2 • Uninhibited proliferation Etiopathogenesis 13
Clinical features A. Symptoms • At diagnosis – 70% symptomatic – Easy fatigability – Loss of sense of well-being – Decreased tolerance to exertion – Anorexia – Abdominal discomfort – Early satiety * – Weight loss – Excessive sweating 15
A. Symptoms • Uncommon symptoms – Bleeding – Night sweats – Heat intolerance – Gouty arthitis – Left upper-quadrant and left shoulder pain* – Urticaria – Hyperleukocytic Syndrome —dyspnea, tachypnea, hypoxia, lethargy, slurred speech Clinical features 16
A. Symptoms Acute febrile neutrophilic dermatosis (Sweet syndrome) Clinical features 17
B. Signs • Pallor • Splenomegaly • Sternal tenderness • Rarely hepatomegaly, lymphadenopathy – Poor prognostic indicators Clinical features 18
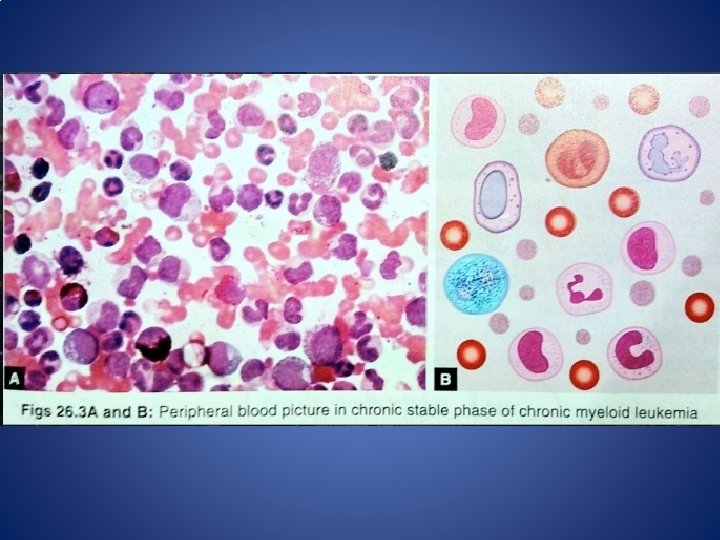
Diagnosis A. Laboratory studies Blood counts and blood smear • Hemoglobin concentration is decreased • Nucleated red cells in blood film • The leukocyte count above 25, 000/μl (even > 1, 000/μl), • Hypersegmented neutrophils • The basophil and eosinophil counts are increased (Absolute) • The platelet count is normal or increased • Blast cells ~ 3 % (<10% in the chronic phase) 19
A. Laboratory studies Peripheral smear Diagnosis 20
A. Laboratory studies Hypercellular Replacement of fat Granulopoiesis Megakaryocytopoiesis Erythropoiesis Bone marrow 21
A. Laboratory studies Bone Marrow studies • Mitotic figures are increased • Macrophages that mimic Gaucher cells * • Macrophages - engorged with lipids - yield ceroid pigment imparting a granular and bluish cast - sea-blue histiocytes. • Dwarf megakaryocytes • Increased reticulin fibrosis (Collagen type III) * • Angiogenesis Diagnosis 22
A. Laboratory studies Other lab features : – Neutrophil Alkaline Phosphatase reduced – Serum B 12 and transcobalamin increased (>10 ULN) – Serum uric acid increased – Lactate dehydrogenase increased – Mean histamine levels increased
B. Cytogenetics • Study of the number and structure of chromosomes • Samples from bone marrow myeloid cells • The presence of the Philadelphia chromosome – shortened chromosome 22* • Cytogenetics cannot identify complex translocations Diagnosis 24
B. Cytogenetics Diagnosis 25
C. Molecular Probes i. FISH (Fluorescence In Situ Hybridization) – Detect the BCR-ABL fusion gene on chromosome 22 – Qualitative Diagnosis 26
C. Molecular Probes ii. PCR (Polymerase Chain Reaction) – Most sensitive test to identify and measure the BCRABL gene (Quantitative) – Can be performed on blood/marrow cells – Amplifies the BCR-ABL derived abnormal m. RNA – One abnormal cell in one million cells can be detected Diagnosis 27
Course of the disease • CML has 3 phases I. Chronic Phase • Most patients are asymptomatic • Incidental leukocytosis/splenomegaly • Bleeding and infectious complications are uncommon in the chronic phase 28
I. Chronic Phase • Most patients are asymptomatic • Incidental leukocytosis/splenomegaly • Bleeding and infectious complications are uncommon in the chronic phase • Usually lasts for 2 to 6 years. • P. smear- NCNC anaemia, Leukocytosis, Blasts <10%, Thrombocytosis. • Decreased LAP score.
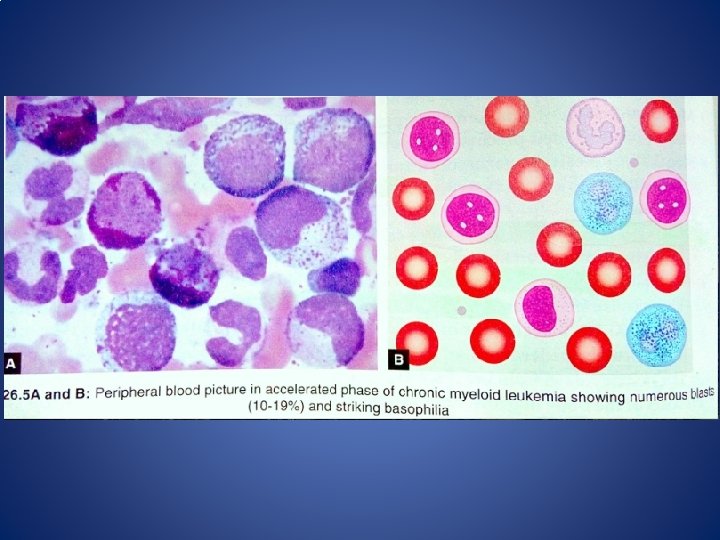
II. Accelerated phase defined by • 10%– 19% blasts in blood or bone marrow • >20% basophils in blood or bone marrow • Thrombocytosis, thrombocytopenia unrelated to therapy (<1 lakh>). • Associated with BM fibrosis. • New clonal chromosome abnormalities • Anemia progresses and cause fatigue, loss of sense of well-being • Splenomegaly • Ranges from 4 -5 years before progressing 31
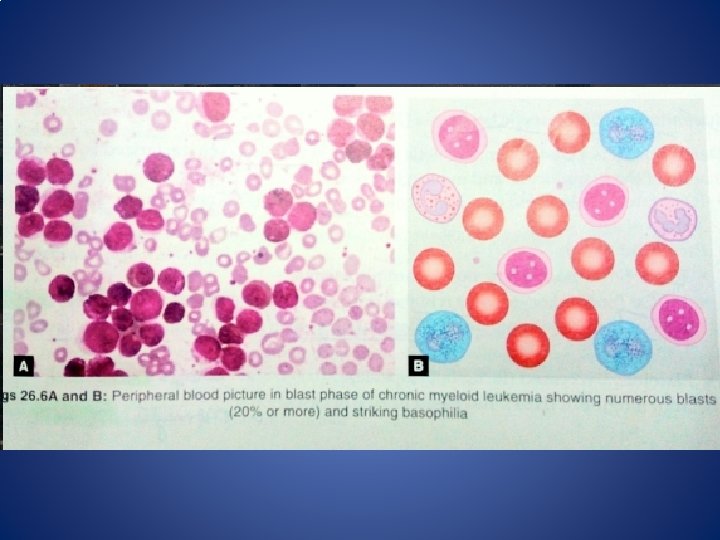
III. Blast Crisis defined by • ≥ 20% blasts in blood or bone marrow • Extramedullary blastic infiltration (Chloroma) – Resembles acute leukemia – 2/3 transform to myeloid blastic phase and 1/3 to lymphoid blastic phase – Infection and bleeding common – Abdominal pain, bone pain – Survival is 6 -12 months (worse for myeloid phenotype) Course of the disease 33
Special Clinical situations Neutrophilic CML – A rare variant of BCR-ABL–positive CML with elevated white cell count principally of mature neutrophils – WBC count lower than that of classic CML – Basophilia, myeloid immaturity in the blood, prominent splenomegaly, or low leukocyte alkaline phosphatase scores – are all absent! – Larger fusion protein than in classic CML – Usually has an indolent course – Now classified separately 35
Special Clinical situations Hyperleukocytosis (15% of cases) – Intravascular flow-impeding effects of white cell counts greater than 3, 000/µL (upto 8 lakh) – Impaired circulation of the lung, central nervous system, special sensory organs, and penis – resulting in some combination of • Tachypnea, dyspnea, cyanosis, • Dizziness, slurred speech, delirium, stupor, • Visual blurring, diplopia, retinal vein distention, retinal hemorrhages, papilledema, • Tinnitus, impaired hearing, • And priapism Course of the disease 36
Special Clinical situations Concurrence of Lymphoid Malignancies 1. CML years after irradiation of non-Hodgkin or Hodgkin lymphoma 2. Accelerated phase dedifferentiation of the CML clone acute lymphoblastic transformation 3. Plasmacytic malignancies – positive association 4. Patients may present with Ph+ ALL, after chemotherapy-induced remission, develop the features of typical CML 37
Differential Diagnosis • • Polycythemia vera Essential thrombocythemia Primary myelofibrosis Leukemoid reactions 38
Treatment 1. 2. 3. 4. 5. 6. 7. 8. Initial Cytoreduction Therapy Tyrosine Kinase Inhibitor Therapy Interferon therapy Chemotherapy Allogeneic Stem Cell Transplantation Treatment of accelerated/blast phases Treatment of CML in pregnancy Treatment cessation 39
1. Initial therapy – Allopurinol 300 mg/day orally with adequate hydration Rasburicase 0. 2 mg/kg i. v (one doses) for Hyperuricemia* – Leukapheresis – helps reduce leucocyte burden, only in conjunction with definitive therapy – Hydroxyurea - Reversible suppression of hematopoiesis 1 to 6 g/day orally (titre based on counts) – Anagrelide – to reduce the platelet burden Treatment 40
2. Tyrosine Kinase inhibitor therapy First generation Second generation Dasatinib Nilotinib Bosutinib Ponatinib Bafetinib Imatinib Treatment 41
Treatment 42
Conclusion • CML- 3 phases: Chronic phase, accelerated phase, blast phase. • Lab P. smear and Bone marrow findings are typical. • Anemia and splenomegaly. • Imatinib has revolutionized the management of CML 43
CML The average at presentation is 45 yrs (rare in children) Death is usually secondary to blast crisis, marrow failure or marrow fibrosis. Symptoms Physical Findings % % Fatigue 80 Splenomegaly 95 Weight loss 60 Hepatomegaly 50 Abdominal fullness 40 Sternal tenderness 80 Easy bruising or hemorrhage 35 Purpura 25 Abdominal pain (splenic infarcts) 30 Retinal hemorrhage 20 Fever, hypermetabolic 10