Prdiction de Structures Inorganiques par Contraintes Gomtriques Armel





 quatre ans plus tôt : \"computational methods can now make detailed")



 : 1 - S. M. Woodley,")
 pour")







 des systèmes chimiques est")

 Développée par Mellot-Draznieks et al. ,")



















")
")
")

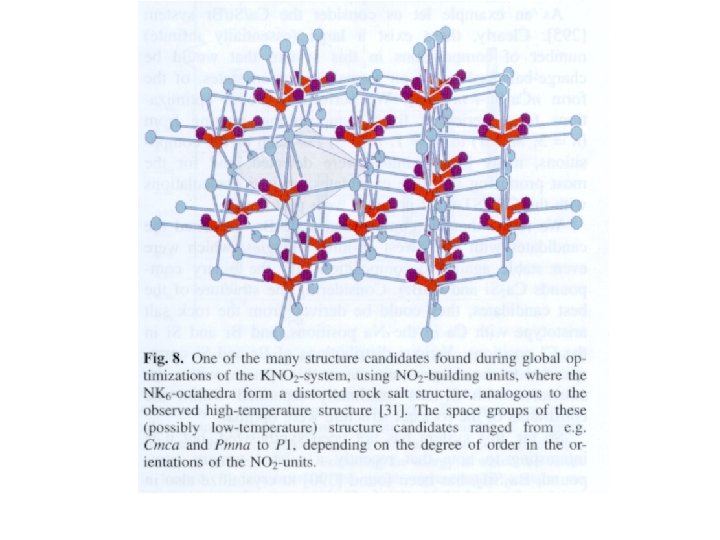
, pas si facile à imaginer, intercroissance pyrochlore-perovskite")

")
.")

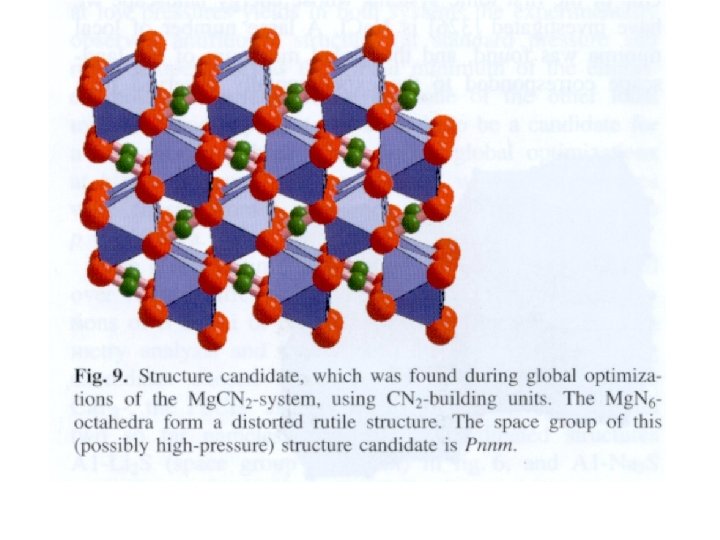
, tetraèdres d’octaèdres, exclusivement")
, tunnels HTB intercroisés at 90° dans le plan ab, pas")
, sans doute pas viable à cause de distorsions élevées des")








 L’exploration des 230 groupes d’espace est réalisée")
![Titanosilicate hypothétique [Si 6 Ti 4 O 22]8 -, R = 0. 0101 SG](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-68.jpg "Titanosilicate hypothétique [Si 6 Ti 4 O 22]8 -, R = 0. 0101 SG")
![Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0335 SG : P](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-69.jpg "Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0335 SG : P")
![Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0109 SG : P](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-70.jpg "Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0109 SG : P")
![Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0044 SG :](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-71.jpg "Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0044 SG :")
![Titanosilicate hypothétique [Si 4 Ti. O 11]2 -, R = 0. 0175 SG :](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-72.jpg "Titanosilicate hypothétique [Si 4 Ti. O 11]2 -, R = 0. 0175 SG :")
![Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0165 SG :](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-73.jpg "Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0165 SG :")
![Titanosilicate hypothétique [Si 6 Ti. O 15]2 -, R = 0. 0124, SG :](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-74.jpg "Titanosilicate hypothétique [Si 6 Ti. O 15]2 -, R = 0. 0124, SG :")


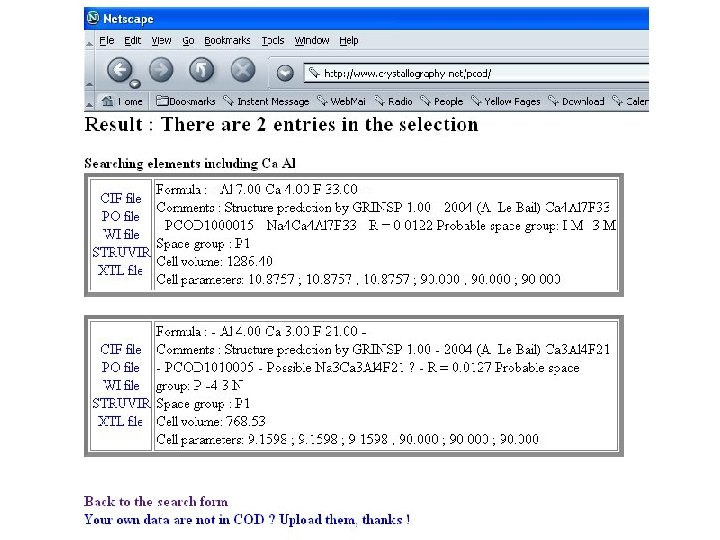
![Proposition virtuelle PCOD 1010005 - [Ca 3 Al 4 F 21]3 -.](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-77.jpg "Proposition virtuelle PCOD 1010005 - [Ca 3 Al 4 F 21]3 -.")
![Proposition virtuelle : [Na. Al 2 F 9]2 -, Groupe d’espace P 4132, a](https://slidetodoc.com/presentation_image/3c7751014ecf76f533593677d6550944/image-78.jpg "Proposition virtuelle : [Na. Al 2 F 9]2 -, Groupe d’espace P 4132, a")




 http: //www. crystallography.")

")
- Slides: 89
Prédiction de Structures Inorganiques par Contraintes Géométriques Armel Le Bail Université du Maine, Laboratoire des oxydes et Fluorures, CNRS UMR 6010, Avenue O. Messiaen, 72085 Le Mans Cedex 9, France. Email : alb@cristal. org
CONTENU - Introduction - Prédiction/Connaissance préalable - Logiciels de Prédiction - GRINSP - Génération des structures - Optimisation des modèles - Composés binaires Polymorphes de B 2 O 3, Si. O 2, Zéolithes, V 2 O 5, Al. F 3 - Sous-produits - Composés ternaires Borosilicates, titanosilicates, fluoroaluminates… - Limitations actuelles - Confirmation des prédictions - Conclusions
INTRODUCTION Prédire une structure cristalline c’est être capable de l’annoncer avant toute synthèse ou découverte à l’état natif. L’intérêt est double : - aider à identifier de nouvelles phases réelles d’après les diagrammes de diffraction de poudre des phases prédites. - sélectionner des phases à synthétiser en fonction de leurs propriétés intéressantes, elles-mêmes prédites. Par ailleurs, un logiciel de prédiction peut aider à déterminer une structure cristalline, mais ce n’est alors plus de la prédiction
Nous sommes très loin de cette chimie prédictive du futur Il faudrait : - des logiciels plus performants de prédiction de structures ET de propriétés (de très gros progrès sont à réaliser), - une base de données de structures prédites suffisamment précises, contenant les coordonnées atomiques, - une base de données de diagrammes de diffraction de poudres de ces structures prédites (pour identification), facile à mettre en œuvre à partir des coordonnées atomiques.
Citation de Frank C. Hawthorne dans un article de fond intitulé: "Structural aspects of oxide and oxysalt crystals" "The goals of theoretical crystallography may be summarized as follow: (1) predict the stoichiometry of the stable compounds; (2) predict the bond topology (i. e. the approximate atomic arrangement) of the stable compounds; (3) given the bond topology, calculate accurate bond lengths and angles (i. e. accurate atomic coordinates and cell dimensions); (4) given accurate atomic coordinates, calculate accurate static and dynamic properties of a crystal. For oxides and oxysalts, we are now quite successful at (3) and (4), but fail miserably at (1) and (2)" F. C. Hawthorne, Acta Cryst. B 50 (1994) 481 -510.
Autre citation (contradictoire) quatre ans plus tôt : "computational methods can now make detailed and accurate predictions of the structures of inorganic materials". C. R. A Catlow & G. D. Price, Nature 347 (1990) 243 -248. Alors, qui croire concernant la prédiction des structures inorganiques ? Le fait est qu’il n’y a pas beaucoup d’exemples de prédiction dans un livre récent édité par C. R. A. Catlow en 1997, excepté des zéolithes. Computer Modelling in Inorganic Crystallography, C. R. A Catlow (ed), Academic Press, 1997. Note: concernant la prédiction des molécules organiques, de récents tests en aveugle démontrent que ce n’est pas très brillant non plus. W. D. S. Motherwell et al. , Acta Cryst. B 58 (2002) 647 -661.
Où en sommes-nous en matière de prédiction de structures inorganiques ? Si l’état de l’art avait sensiblement évolué ces dix dernières années, nous devrions disposer d’énormes bases de données de composés aux structures et propriétés prédites, et aucune nouvelle structure cristalline réelle ne devrait nous surprendre, puisqu’elle devrait correspondre à un jeu de données dans cette banque… D’ailleurs, nous aurions synthétisé de préférence les composés les plus intéressants de cette base. Naturellement, ce n’est absolument pas le cas…
Mais les choses sont peut-être en train de changer : Deux bases de données de structures cristallines de composés hypothétiques ont été créées en 2004 : L’une est exclusivement consacrée aux zéolithes : M. D. Foster & M. M. J. Treacy - Hypothetical Zeolites – http: //www. hypotheticalzeolites. net/ L’autre inclus les zéolithes aussi bien que d’autres oxydes (phosphates, borosilicates, titanosilicates, etc) et des fluorures : A. Le Bail – PCOD : Predicted Crystallography Open Database - http: //www. crystallography. net/pcod/
Prédictions / Connaissance préalables Beaucoup de « prédictions » de structures sont faites alors qu’un composé est déjà synthésisé et que sa composition chimique et ses paramètres de maille sont connus ! Les cas très souvent cités sont ceux des structures pourtant très simples de Li 3 Ru. O 4, ou de Li. Co. F 4 (type di-rutile), de Nb. F 4, etc. Il s’agit clairement alors plutôt de « détermination de structure » que de réelle prédiction. Cependant, il apparaît justifié de faire usage de nos connaissances générales déduites de composés déjà caractérisés (énergies potentielles, distances interatomiques, lien de valence, etc) comme règles de prédiction pour proposer des composés qui restent à découvrir, parce que des prédictions totalement ab initio ne sont pas encore réalisables à grande échelle (les approches par mécanique quantique sont toujours très coûteuses en temps de calcul et ne peuvent pas encore être appliquées pour des explorations systématiques).
Logiciels de prédiction Lectures recommandées (articles de revue) : 1 - S. M. Woodley, in: Application of Evolutionary Computation in Chemistry, R. L. Johnston (ed), Structure and bonding series, Springer. Verlag 110 (2004) 95 -132. 2 - J. C. Schön & M. Jansen, Z. Krist. 216 (2001) 307 -325; 361 -383. Mécanique quantique - HF (Hartree-Fock) : logiciel CRYSTAL (Dovesi et al. , 1989) - DFT (Density Functional Theory) : logiciel CASTEP - DFT + FP-LAPW (Full-Potential linearized augmented plane wave) : logiciel WIEN 2 K
CASTEP Utilise la théorie de la densité fonctionnelle (density functional theory = DFT) pour une modélisation ab initio, en appliquant un code à pseudo-potentiels d’onde plane. M. C Payne et al. , Rev. Mod. Phys. 64 (1992) 1045. Exemple : polymorphes du carbone
Polymorphe hypothétique du carbone suggéré par CASTEP
Une autre prédiction du logiciel CASTEP
Zéolithes Les structures rassemblées dans la base de données des zéolithes hypothétiques sont produites par une grappe de 64 processeurs tournant en continu, générant des modèles, simulant leur recuit. Les réseaux sélectionnés sont ensuite optimisés au moyen du logiciel GULP (General Utility Lattice Program, écrit par by Julian Gale) au moyen de potentiels atomiques. M. D. Foster & M. M. J. Treacy - Hypothetical Zeolites – http: //www. hypotheticalzeolites. net/ > 100000 modèles (non uniques)
Quelques exemples
GULP Ce logiciel est lui-même capable de prédiction de structures (le mode d’emploi décrit un exemple 24 qui fournit les données pour la prédiction des polymorphes de Ti. O 2). Récemment, un algorithme génétique a été introduit dans GULP permettant de générer des charpentes tridimensionnelles à partir de la “seule” connaissance des paramètres de maille et des atomes constitutifs (ce n’est donc plus de la prédiction…), les structures des meilleurs candidats produits sont relaxés par minimisation d’énergie de réseau, basée sur le modèle de Born des solides. S. M. Woodley, in: Application of Evolutionary Computation in Chemistry, R. L. Johnston (ed), Structure and bonding series, Springer-Verlag 110 (2004) 95 -132. GULP : J. D. Gale, J. Chem. Soc. , Faraday Trans. , 93 (1997) 629 -637. http: //gulp. curtin. edu. au/
Partie de la liste des commandes de GULP :
Logiciel G 42 Un concept de “panorama d’énergie” (energy landscape) des systèmes chimiques est proposé par Schön et Jansen pour la prédiction de structures. J. C. Schön & M. Jansen, Z. Krist. 216 (2001) 307 -325; 361 -383.
SPu. DS Un logiciel dédié à la prédiction du mode de tilting des perovskites. M. W. Lufaso & P. M. Woodward, Acta Cryst. B 57 (2001) 725 -738.
Méthode AASBU (Automated Assembly of Secondary Building Units) Développée par Mellot-Draznieks et al. , C. Mellot-Drazniek, J. M. Newsam, A. M. Gorman, C. M. Freeman & G. Férey, Angew. Chem. Int. Ed. 39 (2000) 2270 -2275; C. Mellot-Drazniek, S. Girard, G. Férey, C. Schön, Z. Cancarevic, M. Jansen, Chem. Eur. J. 8 (2002) 4103 -4113. Utilisation de Cerius 2 (commercial) et de GULP dans une séquence de recuit simulé et de minimisation d’énergie pour l’aggrégation de motifs structuraux plus ou moins grands (du polyèdre aux blocs organisés spécifiques de plusieurs polyèdres). Cerius 2, Version 4. 2, Molecular Simulations Inc. , Cambridge, UK, 2000.
Logiciel GRINSP Geometrically Restrained Inorganic Structure Prediction Applique la connaissance des caractéristiques géométriques communes d’un groupe défini de composés (Réseaux 3 D Nconnecté, avec N = 3, 4, 5, 6 et réseaux résultants de la combinaison de 2 valeurs de N), pour la prédiction par un algorithme de type Monte Carlo. Dans GRINSP, la qualité d’un modèle est établie par un facteur R dépendant des écarts pondérés entre distances interatomiques calculées et idéales entre premiers voisins M-X, X-X et M-M dans des composés Ma. Xb ou Ma. M'b. Xc. Ces modèles peuvent nécessiter une optimisation ultérieure au moyen des règles de lien de valence ou par minimisation d’énergie, mais dans la plupart des cas, les paramètres de maille prédits diffèrent de moins de 2% des paramètres observés (cas des structures déjà connues).
Comparaison de quelques paramètres de maille prédits par GRINSP avec des paramètres observés Prédits (Å) Observés ou idéalisés (Å) Dense Si. O 2 Quartz Tridymite Cristobalite Keatite a 4. 965 5. 073 5. 024 7. 525 b 4. 965 5. 073 5. 024 7. 525 c 5. 375 8. 400 6. 796 9. 066 R 0. 0006 0. 0043 0. 0010 0. 0046 a 4. 912 5. 052 4. 969 7. 456 b 4. 912 5. 052 4. 969 7. 456 c 5. 404 8. 270 6. 926 8. 604 Zéolithes ABW EAB EDI GIS GME JBW LTA RHO 9. 872 13. 158 6. 919 9. 772 13. 609 5. 209 11. 936 14. 926 5. 229 13. 158 6. 919 9. 772 13. 609 7. 983 11. 936 14. 926 8. 733 15. 034 6. 407 10. 174 9. 931 7. 543 11. 936 14. 926 0. 0056 0. 0037 0. 0047 0. 0027 0. 0031 0. 0066 0. 0035 0. 0022 9. 9 13. 2 6. 926 9. 8 13. 7 5. 3 11. 9 14. 9 5. 3 13. 2 6. 926 9. 8 13. 7 8. 2 11. 9 14. 9 8. 8 15. 0 6. 410 10. 2 9. 9 7. 5 11. 9 14. 9 Fluorures d’aluminium -Al. F 3 10. 216 Na 4 Ca 4 Al 7 F 33 10. 860 Al. F 3 -pyrochlore 9. 668 10. 216 10. 860 9. 668 7. 241 10. 860 9. 668 0. 0162 0. 0333 0. 0047 10. 184 10. 781 9. 749 7. 174 10. 781 9. 749
Plus de détails sur l’algorithme dans GRINSP Etape 1 : Génération des structures Des atomes M/M’ sont placés au hasard l’un après l’autre dans une maille dont les dimensions sont sélectionnées également au hasard. A mesure que le remplissage s’effectue, le modèle doit respecter exactement les spécifications géométriques (nombres de voisins M/M’ exacts au final du remplissage, mais une large tolérance est accordée sur les distances M-M, M-M’ et M’-M’). Cette large tolérance sur les distances permet la capture de solutions qui ne correspondent pas toujours à des polyèdres réguliers. A cette première étape les atomes M/M’ ne bougent pas, leurs positions possibles sont testées puis retenues ou non. La maille est progressivement remplies jusqu’à ce que les restrictions géométriques soient vérifiées, si possible. Le nombre d’atomes M/M’ placés n’est pas prédéterminé.
Etape 2 : optimisation des modèles Les atomes X sont ajoutés à mi-chemin des (M/M')-(M/M') premiers voisins (liaisons par sommets) et il est vérifié par variations Monte Carlo des positions et des paramètres de maille que des polyèdres réguliers (M/M’)Xn peuvent réellement être proposés. La fonction de coût est basée sur la vérification de distances interatomiques fournies M-M, M-X et X-X idéales entre premiers voisins. Un facteur R total est défini comme : R = [(R 1+R 2+R 3)/ (R 01+R 02+R 03)], où Rn et R 0 n pour n = 1, 2, 3 correspondent à : Rn = [wn(d 0 n-dn)]2, R 0 n = [wnd 0 n]2, où d 0 n sont les distances interatomiques idéales M-X (n=1), X-X (n=2) and MM (n=3), tandis que dn sont les distances observées dans le modèle structural. Les poids attribués (wn) proviennent du logiciel DLS (Distance Least Square) pour le calcul des charpentes de zéolithes idéalisées (w 1= 2. 0, w 2 = 0. 61 et w 3 = 0. 23).
Cette minimisation des écarts de distances observées/idéales est une approche très élémentaire du problème… Les distances idéales doivent être fournies par l’utilisateur du logiciel, pour des paires d’atomes supposés former des polyèdres. Par exemple dans le cas de tétraèdres [Si. O 4] connectés par sommets, les distances idéales sont d 1 = 1. 61 Å (Si-O), d 2 = 2. 629 Å (O-O) et d 3 = 3. 07 Å (Si-Si). Pour des composés ternaires, les distances idéales M-M' sont calculées dans GRINSP comme la moyenne des distances idéales M-M et M'-M' fournies par l’utilisateur. Un défaut est de ne considérer, pour le calcul de R, que des distances X-X intra-polyèdres, négligeant les distances X-X inter-polyèdres. Ce facteur R pourrait être mieux défini différemment, par exemple au moyen des règles de lien de valence (en projet pour une prochaine version de GRINSP). Cette approche élémentaire ne fonctionne bien que pour des polyèdres réguliers
Plus de détails sur l’étape d’optimisation Durant cette seconde étape, les atomes M/M’/X bougent, mais légèrement, sans jamais rompre les liaisons. Les paramètres de maille du modèle de départ sont aussi modifiés dans le but de recher le minimum pour le facteur R. Ces paramètres peuvent finir par varier considérablement par rapport à ceux du modèle initial (jusqu’à 30%). En moyenne 20000 tests Monte Carlo sont effectuées par atome du modèle. Pendant cette optimisation, le groupe d’espace original utilisé pour placer les atomes M/M' peut changer après que les atomes X aient été positionnés. La structure finale est toujours proposée dans le groupe d’espace P 1, et présentée sous forme de ficher CIF. Le choix final de la symétrie réelle doit être fait au moyen d’un logiciel tel que PLATON (A. Spek).
Composés binaires prédits par GRINSP Les formulations M 2 X 3, MX 2, M 2 X 5 et MX 3 à polyèdres connectés exclusivement par sommets ont été en grande partie examinées (correspondant respectivement à N = 3, 4, 5, 6). Zéolithes (MX 2) – Une exploration complète n’est pas terminée. Plus de 1000 modèles ont été produits par GRINSP, incluant > 50 structures connues, pour R < 0. 01, des paramètres de maille < 16 Å et un nombre maximum de 64 Si par maille. GRINSP reconnait un zéotype par comparaison des séquences de coordination (CS) d’un modèle avec les CS déjà obtenus et stockés dans une liste (ainsi qu’avec ceux déjà rencontrés pendant l’exécution en cours du logiciel). Les fichiers CIF peuvent être consultés dans la base de donnée PCOD, en précisant le numéro de code de l’entrée (par exemple PCOD 1010026, etc), ou paramètres de maille, volume, etc.
Zéolithe hypothétique PCOD 1010026 SG : P 432, a = 14. 623 Å, FD = 11. 51
Zéolithe hypothétique PCOD 1010038 SG : P 432, a = 14. 70 Å - FD = 11. 32 formulation : [Si 2 Al. O 6]-1
Zéolithe hypothétique PCOD 1030060 SG : P 63 mc, a = 18. 74 Å, c = 9. 02Å, FD = 14. 6.
Zéolithe hypothétique PCOD 1030081 SG : P 6/m, a = 15. 60 Å, c = 7. 13Å, FD = 16. 0.
Polymorphes hypothétiques de B 2 O 3 prédits par GRINSP Il n’existe que peu de variétés connues de B 2 O 3. GRINSP en propose un nombre considérable. . . Hypothétique B 2 O 3 PCOD 1062004.
Hypothétique B 2 O 3 PCOD 1051002
Polymorphes de Al. F 3 prédits par GRINSP (réseaux 3 D 6 -connectés : octaèdres liés exclusivement par sommets) La structure de type perovskite peut être prédite par GRINSP dans presque tous les groupes d’espace, et toutes les autres variétées connues (type pyrochlore, HTB, TTB) sont retrouvées par le logiciel, incluant la toute dernière découverte en 1992, -Al. F 3. Des polymorphes inconnus pour Al. F 3 mais existant pour d’autres formulations MX 3, avec cations intersticiels ou non, sont également prédits par GRINSP. Enfin, GRINSP suggère qu’il resterait encore des possibilités de découverte de nouvelle formes…
Résumé des conditions de prédiction des variétés de Al. F 3 - Recherche complète dans les 230 groupes d’espace, mais pour des paramètres de maille inférieurs à 16 Å et moins de 64 Al par maille. - Distances idéales entre premiers voisins : 3. 5 Å pour Al-Al, 1. 81 Å pour Al-F et 2. 559 Å pour F-F (octaèdres parfaits, liaison légèrement tiltée) dans le calcul du facteur R. - De 10000 à 200000 tests indépendants par groupe d’espace (tests pour des mailles différentes). - Jusqu’à 300000 évènements Monte Carlo dans chaque test (pour positionner les atomes d’aluminium). - De 10000 à 20000 évènements Monte Carlo à l’étape de l’optimisation (déplaçant légèrement Al ou F, ou modifiant les paramètres de maille pour atteindre une valeur de R minimale). - 24 heures de calcul par groupe d’espace sur un processeur à 2. 6 GHz.
Classification des polymorphes de Al. F 3 proposés par GRINSP (identifiés comme connus ou inconnus) en fonction de R croissant Structure-type FD a b c SG Z N R HTB 19. 68 6. 987 7. 212 90. 00 120. 00 P 63/mmc 6 1 0. 0035 Tl. Ca 2 Ta 5 O 15 20. 67 7. 004 7. 228 9. 558 90. 00 Pmmm 10 2 0. 0040 U-1 (Al. F 3) 21. 27 6. 992 7. 218 13. 513 90. 00 105. 22 90. 00 P 21/m 14 3 0. 0042 Pyrochlore 17. 71 9. 668 90. 00 Fd-3 m 16 1 0. 0046 U-2 (Al. F 3) 20. 43 6. 889 8. 252 90. 00 P-4 m 2 8 2 0. 0057 Perovskite 21. 16 3. 615 90. 00 Pm-3 m 1 1 0. 0063 Ba 4 Co. Ta 10 O 30 21. 15 9. 499 13. 777 7. 224 90. 00 Iba 2 20 2 0. 0095 TTB 20. 78 11. 539 7. 229 90. 00 P 42/mbc 20 2 0. 0099 U-3 (Al. F 3) 22. 37 6. 960 7. 402 5. 207 90. 00 Pnc 2 6 2 0. 0160 -Al. F 3 21. 17 10. 214 7. 242 90. 00 P 4/nmm 16 3 0. 0162 U-4 (Al. F 3) 21. 71 10. 505 6. 678 90. 00 I 41/a 16 1 0. 0181 U-5 (Al. F 3) 19. 74 7. 125 11. 977 90. 00 P 42/mmc 12 2 0. 0191 U-6 (Al. F 3) 23. 65 12. 601 6. 391 90. 00 P 4/nmm 12 2 0. 0233 U-7 (Al. F 3) 19. 22 6. 396 5. 087 90. 00 P 42 mc 4 1 0. 0243 U-8 (Al. F 3) 19. 65 10. 624 7. 212 90. 00 P 4/mmm 16 2 0. 0275 Na 4 Ca 4 Al 7 F 33 23. 27 9. 805 9. 834 90. 00 I 4/mmm 22 3 0. 0283 U-9 (Al. F 3) 23. 46 7. 997 90. 00 P 4132 12 2 0. 0287 U-10 (Al. F 3) 17. 68 6. 874 14. 360 90. 00 P 42 mc 12 2 0. 0299 FD = framework density (= densité du squelette = nombre d’atomes Al atoms pour un volume de 1000Å 3). SG = groupe d’espace de plus haute symmétrie dans lequel le modèle initial des seuls atomes d’Al a été obtenud (pas nécessairement le groupe d’espace final après positionnement des atomes de fluor). Z = nombre de formules Al. F 3 par maille. N = nombre d’atomes Al présentant des séquences de coordination différentes. R = facteur de qualité basé sur le respect des distances idéales entre premiers voisins Al-F, F-F etd Al-Al.
Type structural HTB (Hexagonal Tungsten Bronze)
Type structural Tl. Ca 2 Ta 5 O 15 , Intercroissance HTB-perovskite (2 plans)
Modèle inconnu U-1, mais simple à imaginer : intercroissance HTB-perovskite (3 plans)
Structure-type pyrochlore, construit à partir de tétraèdres d’octaèdres
Variété inconnue U-2 (Al. F 3), pas si facile à imaginer, intercroissance pyrochlore-perovskite
Structure-type Ba 4 Co. Ta 10 O 30
Type structural TTB (Tetragonal Tungsten Bronze)
Variété inconnue U-3 (Al. F 3).
Polymorphe connu -Al. F 3 - tetraèdres et chaines d’octaèdres
U-4 (Al. F 3), tetraèdres d’octaèdres, exclusivement
U-5 (Al. F 3), tunnels HTB intercroisés at 90° dans le plan ab, pas si facile à imaginer…
U-6 (Al. F 3), sans doute pas viable à cause de distorsions élevées des octaèdres et de distances F-F inter-octaèdres trop courtes (R > 0. 02)
Sous-produits des prédictions du logiciel GRINSP Par exemple, des polyèdres à 6 sommets autres qu’un octaèdre peuvent être produits : prismes trigonaux ou pyramides à base pentagonale. Mais comme ils ne sont pas réguliers (pas de distance idéale unique F-F ou Al -F distance), les structures proposées par GRINSP sont classées avec un facteur R élevé. Dans les fluorures connus, l’aluminium est toujours en coordination octaèdrique, ces prédictions avec d’autres polyèdres sont donc très certainement irréalistes, mais certains cations dans les oxydes ou même fluorures adoptent ces autres formes polyèdriques. D’un point de vue esthétique, ces autres prédictions sont intéressante, d’autant plus que certaines structures ont des densités de squelette – ou de charpente - (framework density = FD) faibles, montrant des cavités ou tunnels énormes qui les rendent attractives.
Octaèdres + pyramides à base pentagonale :
Octaèdres + prismes trigonaux :
De plus, si la majorité des modèles qui respectent la condition géométrique du partage des sommets des polyèdres sont tridimensionnels, certains sont bidimensionnels (structures en couches). Pour ces cas-là, GRINSP ne dispose d’aucun moyen pour estimer correctement les distances entre couches. Des modèles mono-dimensionnels peuvent même être proposés. Ici, des cylindres de formulation B 2 O 3. De même, les distances entre ces cylindres ne sont pas évaluées correctement par GRINSP :
GRINSP permet aussi d’explorer les composés ternaires à charpentes de polyèdres à sommets communs On considère ici deux sortes de cations, M et M’. Ils peuvent avoir soit : - une même coordination mais des rayons ioniques différents, - une coordination différente. En conséquence, seulement certaines formulations peuvent être produites et de plus, si M ou M' ne forment pas des composés binaires électriquement neutres, alors les ternaires ne seront pas neutres non plus. Tous les borosilicates proposés par GRINSP sont automatiquement électriquement neutres, mais pas les aluminophosphates ou les titanosilicates, etc.
Borosilicates virtuels Il n’existe qu’un seul borosilicate répertorié dans la base ICSD. GRINSP en propose de grandes quantités, même en limitant à R < 0. 006, 57 modèles différents avec la formulation Si. B 2 O 5 32 modèles pour Si 3 B 4 O 12 28 pour Si 2 B 6 O 13 et Si 4 B 2 O 11 24 pour Si 2 B 2 O 7, 18 pour Si. B 6 O 11, 17 pour Si. B 4 O 8, 14 pour Si 3 B 2 O 9, 6 pour Si 6 B 2 O 15 et 2 for Si 3 B 6 O 15. Plus 369 autres modèles non classés en symétrie triclinique ! Et encore, l’étude n’est pas réalisée pour tous les groupes d’espace…
PCOD 2050102, Si 5 B 2 O 13, R = 0. 0055.
PCOD 2050018, Si 3 B 4 O 12, R = 0. 0039.
Titanosilicates hypothétiques (à octaèdres/tetraèdres à sommets communs) L’exploration des 230 groupes d’espace est réalisée mais le dépouillement des résultats n’est pas terminé. Certains modèles ne sont pas forcément viables si les polyèdres n’acceptent pas une certaine distorsion (modèles avec R > 0. 02). Ces modèles ne sont pas électriquement neutres, et donc les charpentes doivent pouvoir disposer de sites intersticiels pour des cations ou molécules afin d’exister éventuellement. Une prochaîne étape du développement du logiciel GRINSP est donc clairement cet équilibrage électrique par ajout automatique de cations ou molécules chargées dans les sites intersticiels ou dans les grosses cavités ou les tunnels. Mais alors, de simples critères géométriques de construction ne suffisent plus, il faut des considérations de lien de valence et d’energie de réseau.
Titanosilicate hypothétique [Si 6 Ti 4 O 22]8 -, R = 0. 0101 SG : P-4 m 2, a = 7. 564 Å, c = 9. 702 Å, FD = 16. 2.
Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0335 SG : P 42/mmc, a = 12. 85 Å, c = 7. 76 Å, FD = 12. 5.
Titanosilicate hypothétique [Si. Ti. O 5]2 -, R = 0. 0109 SG : P 42/mmc, a = 10. 47 Å, c = 7. 64 Å, FD = 19. 1.
Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0044 SG : P 42/mmc, a = 7. 73 Å, c = 10. 50 Å, FD = 19. 1.
Titanosilicate hypothétique [Si 4 Ti. O 11]2 -, R = 0. 0175 SG : P 42/mmc, a = 12. 89 Å, c = 9. 23 Å, FD = 13. 0.
Titanosilicate hypothétique [Si 2 Ti. O 7]2 -, R = 0. 0165 SG : P 42/nbc, a = 12. 32 Å, c = 7. 27 Å, FD = 21. 7.
Titanosilicate hypothétique [Si 6 Ti. O 15]2 -, R = 0. 0124, SG : P 63, a = 11. 97 Å, c = 6. 30 Å, FD = 17. 9.
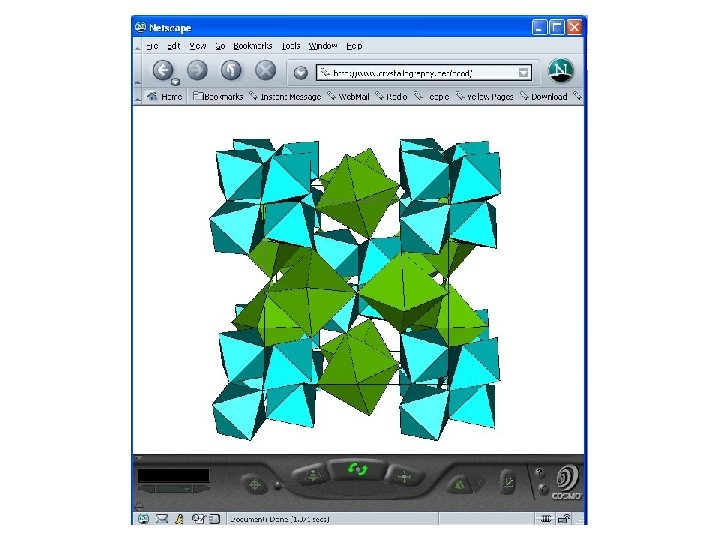
Fluoroaluminates Cette catégorie de composés a été seulement en partie explorée au moyen du logiciel GRINSP, combinant des octaèdres de tailles différentes ([Al. F 6] and [Ca. F 6] or [Na. F 6]). Quelques réseaux mixtes connus (3 D 6 connectés) ont été retrouvés comme [Ca 4 Al 7 F 33]4 - existant avec pour formulation Na 4 Ca 4 Al 7 F 33. Des charpentes hypothétiques qui pourraient être viables ont été proposées par GRINSP, telle que [Ca 3 Al 4 F 21]3 - ou [Na. Al 2 F 9]2 -.
Connu dans Na 4 Ca 4 Al 7 F 33 PCOD 1000015 - [Ca 4 Al 7 F 33]4 -.
Proposition virtuelle PCOD 1010005 - [Ca 3 Al 4 F 21]3 -.
Proposition virtuelle : [Na. Al 2 F 9]2 -, Groupe d’espace P 4132, a = 9. 05Å.
Rappel des limitations du logiciel GRINSP Exploration limitée aux réseaux 3 D et 3 -, 4 -, 5 - ou 6 -connectés, produisant des polyèdres connectés par sommets pour des composés binaires (M 2 X 3, MX 2, M 2 X 5 ou MX 3) ou ternaires (Ma. Mb'Xc). Extensions envisagées Il est concevable de modifier l’algorithme pour permettre des prédictions de composés à liaisons par arêtes et/ou faces et/ou sommets, avec remplissage automatique des cavités/tunnels pour parvenir à une neutralité électrique. Une utilisation des règles de lien de valence aurait une portée plus générale que de simples restrictions géométriques pour l’optimisation des modèles.
La question de la confirmation des prédictions Une prédiction de formule simple comme Si. O 2 or Al. F 3 ne donne pas assez d’indications, mais si la formulation est ternaire ou quaternaire, il est au moins possible d’essayer les méthodes classique de synthèse à l’état solide. Si un modèle intéressant est prédit pour une formulation [Ca 3 Al 4 F 21]3 -, on peut envisager de tenter de synthétiser Na 3 Ca 3 Al 4 F 21 ou Li 3 Ca 3 Al 4 F 21. Certainement, la plupart des prédictions seront vaines, jamais vérifiées, même pour des phases vraiment viables, parce que la synthèse peut dépendre d’un précurseur organométallique, ou d’un hydrate, ou d’un composé amorphe qui lui-même est encore inconnu…
Par exemple Une variété de fluorure découverte en 1992, -Al. F 3 est synthétisée par décomposition thermique soit de [(CH 3)4 N]Al. F 4 H 2 O ou bien d’un amorphe de composition Al. F 3 x. H 2 O (x < 0. 5). C’est un exemple unique, aucun autre MX 3 n’adopte ce type structural, bien qu’il n’y ait aucune objection géométrique ou de nature énergétique à l’existence possible de phases analogues -Fe. F 3, -VF 3 or -Cr. F 3. L’existence d’une base de données de structures prédites aurait grandement facilité la détermination de la structure de -Al. F 3 qui n’existe que sous forme de poudre.
CONCLUSION La prédiction des structures et de leurs propriétés apparaît être un sujet de recherche important et inévitable du futur de la cristallographie et de la chimie inorganique (et organique ou organométallique…).
Pour finir Quelques copies d’écran de PCOD (Predicted Crystallography Open Database) http: //www. crystallography. net/pcod/ Une base de données qui souhaite recueillir vos prédictions avec GRINSP ou d’autres logiciels.
PCOD est un sous-ensemble de COD contenant les données cristallographiques de composés réels
GRINSP est un logiciel sous licence GPL ( « Open source » )