Genome sequence assembly concepts and methods ShihJon Wang

Genome sequence assembly concepts and methods Shih-Jon Wang May 13, 2008

OUTLINE • • • Assembly Process Overview Assembly algorithms Repeats Scaffolding Phred/Phrap/Consed Assembly pipelines

Assembly process overview

A Genome Sequencing Project
")
Building a Library • Break DNA into random fragments (810 x)

SHOTGUNs • • • Whole Genome Shotgun Bac-Bac Shotgun Size of inserts: --Bac insert: ~150 KB --Fosmid insert: ~30 KB --Normal insert: ~3 KB
 Clone inserts are sequenced from both ends, yielding mated sequence")
Clone and scaffold (a) Clone inserts are sequenced from both ends, yielding mated sequence reads. (b) A scaffold uses linking information provided by the clone-pairing data to order and orient contiguous sequences, or contigs, in the genome under assembly. Computer 35 (7): 47 -54
 -- Amplify the")
Building a Library • Break DNA into random fragments (~10 x) -- Amplify the fragments in a vector -- Sequence 800 -1000 bases at each end

Assembling the fragments

Assembling the fragments • Break DNA into random fragments • Sequence the ends of the fragments • Assemble the sequenced ends

Forward-reverse constraints • The sequenced ends are facing towards each other • The distance between the two fragments is known

Building Scaffolds

Assembly Gaps --sequencing gap: know the order & orientation of the contigs and have at least one clone spanning the gap --physical gap: no information about adjacent contigs, nor about the DNA spanning the gap

Finishing the Project

Unifying View of Assembly

Assembly Algorithms
 –")
Assembly Methods • Overlap-layout-consensus – greedy (Phrap, CAP 3, TIGR. . . ) – graph-based (Euler)

Phrap/CAP 3 Greedy • Build a rough map of fragment overlaps • Pick the largest scoring overlap • Merge the two fragments • Repeat until no more merges can be done !!! IDEAL CASE !!!

Real World Problems • • • Sequencing errors Chimera Repeats Contaminants Polymorphism Orientation

Error Correction

Overlap b/w two sequences

All pairs alignment • Try all pairs – must consider ~ n^2 pairs • Smarter solution: only n x coverage (e. g. 8) pairs are possible – Build a table of k-mers contained in sequences (single pass through the genome) – Generate the pairs from k-mer table (single pass through k-mer table)

Repeats

Repeat sequence The top represents the correct layout of three DNA sequences. The bottom shows a repeat collapsed in a misassembly. Computer 35 (7): 47 -54
, eg. Alu <300 bp Long interspersed")
重覆序列 ■重覆頻率分 Interspersed repeats Short interspersed element (SINE), eg. Alu <300 bp Long interspersed element (LINE), ca. 5 kb Tandem repeats Satellite DNA Minisat. & Variable number of tandem repeats Microsat. : mono-, di-, tri-, tetra-nucleotide ■重覆方向分 同向重覆序列 反向重覆序列
 • repeat")
Repeat detection Pre-assembly: find fragments that belong to repeats • statistically (Reps) • repeat database (Repeat. Masker)

Statistical repeat detection • Significant deviations from average coverage flagged as repeats. - frequent k-mers are ignored - “arrival” rate of reads in contigs compared with theoretical value (e. g. , 800 bp reads & 8 x coverage - reads "arrive" every 100 bp) • Problem 1: assumption of uniform distribution of fragments - leads to false positives non-random libraries poor clonability regions • Problem 2: repeats with low copy number are missed

Scaffolding

Sequencing hierarchy • Random sequencing – unrelated reads ~700 pairs • Assembly – un-related contigs 5 K-10 K pairs • Scaffolding – unrelated scaffolds 30 K~ 50 K pairs • Finishing/gap closure – completed genomes millions-billions of base-pairs

Definition

Scaffolder output • order and orientation of contigs • size of gaps between contigs • linking evidence: mate-pairs spanning gaps

Clone-mates

Linking information

Hierarchical scaffolding

Ambiguous scaffold

Phred/Phrap/Consed Analysis

What is Phred/Phrap/Consed ? Phred/Phrap/Consed is a worldwide distributed package for: a. Trace file (chromatograms) reading; b. Quality (confidence) assignment to each individual base; c. Vector & repeat sequences identification and masking; d. Sequence assembly; e. Assembly visualization and editing; f. Automatic finishing.

How to deal with the enormous amount of reads generated by the high throughput DNA sequencers?

Phred Genome Research 8: 175 -194

Phred is a program that performs several tasks: a. Reads trace files – compatible with most file formats: SCF (standard chromatogram format), ABI, ESD (Mega. BACE) and LI-COR. b. Calls bases – attributes a base for each identified peak with a lower errorrate than the staard base calling programs.

Phred c. Assigns quality values to the bases – a “Phred value” based on an error rate estimation calculated for each individual base. d. Creates output files – base calls and quality values are written to output files.

File Directories • chromat_dir/ • edit_dir/ • phd_dir/
")
Trace File High quality region – no ambiguities (Ns)
")
Trace File Medium quality region – some ambiguities (Ns)

Trace File Poor quality region – low confidence
 where q -")
Phred value formula q = - 10 x log 10 (p) where q - quality value p - estimated probability error for a base call Examples: q = 20 means p = 10 -2 (1 error in 100 bases) q = 40 means p = 10 -4 (1 error in 10, 000 bases)

Base Calling • phred -id. -p -pd. . /phd_dir • phred -view pf 84 c 05. s 1

The structure of a phd file BEGIN_SEQUENCE 01 EBV 10201 A 02. g BEGIN_COMMENT CHROMAT_FILE: EBV 10201 A 02. g ABI_THUMBPRINT: PHRED_VERSION: 0. 990722. g CALL_METHOD: phred QUALITY_LEVELS: 99 TIME: Thu May 24 00: 18: 58 2001 TRACE_ARRAY_MIN_INDEX: 0 TRACE_ARRAY_MAX_INDEX: 12153 TRIM: CHEM: term DYE: big END_COMMENT BEGIN_DNA t 8 5 c 13 17 a 19 26 c 19 32 t a a a g c c t g g t g c c t a a t g 24 24 22 27 25 19 12 15 19 23 33 36 44 44 39 39 34 35 34 2221 2232 2245 2261 2272 2286 2302 2314 2324 2331 2346 2363 2378 2390 2404 2419 2433 2446 2460 2470 2482 t g t c g n c t t c c c t c g g a g g 16 8191 t 6 11908 19 8200 a 6 11921 13 8211 g 6 11927 13 8229 t 6 11947 4 8241 c 6 11953 4 8253 a 6 11964 4 8263 g 6 11981 10 8276 c 4 11994 9 8286 n 4 12015 c 4 12037 12 8301 n 4 12044 16 8313 n 4 12058 12 8329 n 4 12071 12 8336 n 4 12085 15 8343 n 4 12098 19 8356 n 4 12111 9 8371 n 4 12124 13 8386 c 4 12144 14 8397 n 4 12151 7 8417 END_DNA 9 8427 4 8445 END_SEQUENCE

phd 2 fasta • phd 2 fasta program – –converts. phdfiles to sequence in multifasta format – –writes. qualfile (quality scores) for each trace file – –phd 2 fasta -id. . /phd_dir -os CLONE. fasta oq CLONE. fasta. qual • Output: – –fasta. seqcontains fastasequences – –fasta. seq. qualcontains quality scores
 • DNA sequence cleaning: quality trimming and vector removal---Lucy: •")
Vector Sequence Cleaning (1) • DNA sequence cleaning: quality trimming and vector removal---Lucy: • Lucy Steps: – Read input seq#, seq info, and quality info – Chop off splice sites – Remove vector insert – Produce output seq for fragment assembly.
 • Restriction on file name: can’t contain any symbol eg.")
Vector Sequence Cleaning (2) • Restriction on file name: can’t contain any symbol eg. “–” “. “ “_” • Lucy major parameters to set up: -vector_complete. Seq splice_site_file (splice_site_file: 2 splice-site seq before and after the insertion point on the vector) • Lucy Output: – identified locations of good/clean region – trim seq without vector, linker, Ns (<3% Ns)

splice_site_file • ~ 150 bases, 50 bases overlap around splice • >PUCsplice. for. begin gattaagttgggtaacgccagggttttcccagtcacgacgttgtaaaacg acggccagtgccaagcttgcatgcctgcaggtcgactctagaggatcccc gggtaccgagctcgaattcgtaatcatggtcatagctgtttcctgtgtga • >PUCsplice. for. end acggccagtgccaagcttgcatgcctgcaggtcgactctagaggatcccc gggtaccgagctcgaattcgtaatcatggtcatagctgtttcctgtgtga aattgttatccgctcacaattccacacaacatacgagccggaagcataaa • >PUCsplice. rev. begin tttatgcttccggctcgtatgttgtgtggaattgtgagcggataacaatt tcacacaggaaacagctatgaccatgattacgaattcgagctcggtaccc ggggatcctctagagtcgacctgcaggcatgcaagcttggcactggccgt • >PUCsplice. rev. end tcacacaggaaacagctatgaccatgattacgaattcgagctcggtaccc ggggatcctctagagtcgacctgcaggcatgcaagcttggcactggccgt cgttttacaacgtcgtgactgggaaaaccctggcgttacccaacttaatc

Cross_match

Cross_match • cross_match -minmatch 12 -penalty -2 minscore 20 -screen CLONE. fasta /net/share/sequence_pipeline/vector. fasta
 • Phrap is a")
Phrap -- Phragment Assembly Program (or Phil’s Revised Assembly Program) • Phrap is a program for assembling shotgun DNA sequence data • Key Features: • a. Uses the entire read content – no need for trimming. • b. User supplied (i. e. Repbase) + internally computed data – better accuracy of assembly in the presence of repeats. • c. Contig sequence is constituted by a mosaic of the highest quality parts of the reads – it’s not a consensus!

Phrap --Phrap is a program for assembling shotgun DNA sequence data • d. Provides extensive information about assembly – contained in phrap. out, *. ace and *. screen. contigs. qual files • e. Handles very large datasets – hundreds of thousands of reads are easily manipulated. • f. Generate output files – contain some important data and enable visualization by other programs

Banded Search

K-mers • >GL 1234. b 1 gattaagttgggtaacgccagggttttcccagtcac… gattaagttgggtaa ttaagttgggtaacg. . .

Phrap output files • *. contigs – fasta file containing the contigs – Contigs with more than one read – Singletons (single reads with a match to some other contig but that couldn’t be merged consistently to it) • *. singlets – fasta file of the singlet reads – Reads with no match to other read • *. ace – allows for viewing the assembly using Consed • *. view – required for viewing the assembly using Phrapview

Phrap parameters • phrap -new_ace CLONE. fasta. screen >outfile • OPTIONS DEFAULT FUNCTION • -penalty -2 ↑=>↑Stringency. -gap_init penalty -2. -gap_ext penalty -1 • -minmatch* 14 ↑=>↓time↓Matches • -bandwidth 14 ↓=>↓time↓String. • -minscore 30 ↑=>↑String. • *highly sensitive! bigger genomes bigger value

Phrap parameters • • OPTIONS DEFAULT FUNCTION -forcelevel 0~10 ↓=>↑String. -repeat_stringency 0. 95 0<x<1 ↑=>↑String. -force_high* ↑=>↑String. -revise_greedy** ↓Misassembly -shatter_greedy** ↓Contig. Length * Ignore edited high-quality discrepancies **break assembly at weak joins

Phrap parameters • • • OPTIONS DEFAULT -max_subclone_size 5000 -default_qual 15 -preassemble* -group_delim* _ *used together FUNCTION F. -R. check

Consed Genome Research 8: 195 -202, 1998

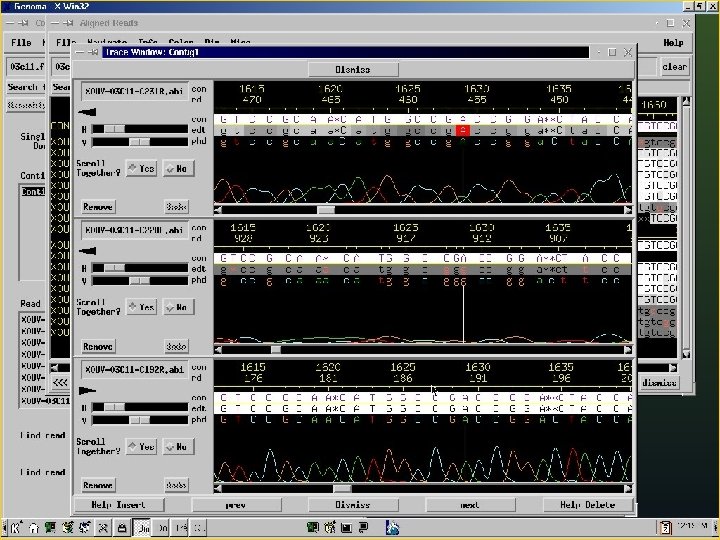
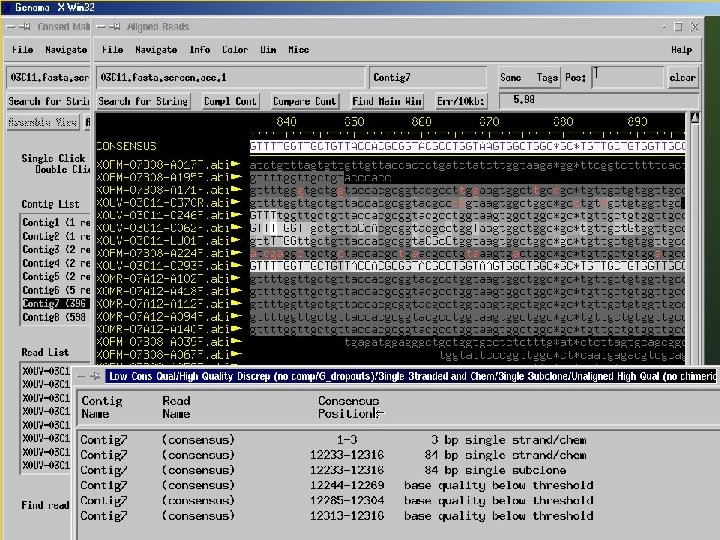
Consed A program for viewing and editing assemblies produced by Phrap Key Features: a. Assembly viewer - allows for visualization of contigs, assembly (aligned reads), quality values of reads and final sequence. b. Trace file viewer – single and multiple trace files can be visualized allowing for comparison of a given sequence in several reads.

Consed A program for viewing and assemblies produced by Phrap editing Key Features: c. Navigation – identify and list regions which are below a given quality threshold, contain high quality discrepancies, single-strand coverage, etc. d. Autofinish – automatic set of functions for: gap closure, improvement of sequence quality, determination of relative orientation of contigs, identification of regions covered by a single read or by reads of a single strand. The program automatically performs primer picking and chooses the templates.
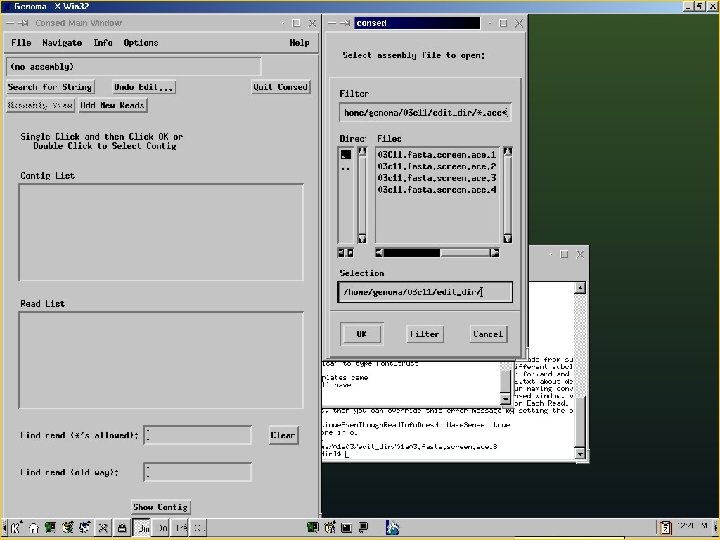
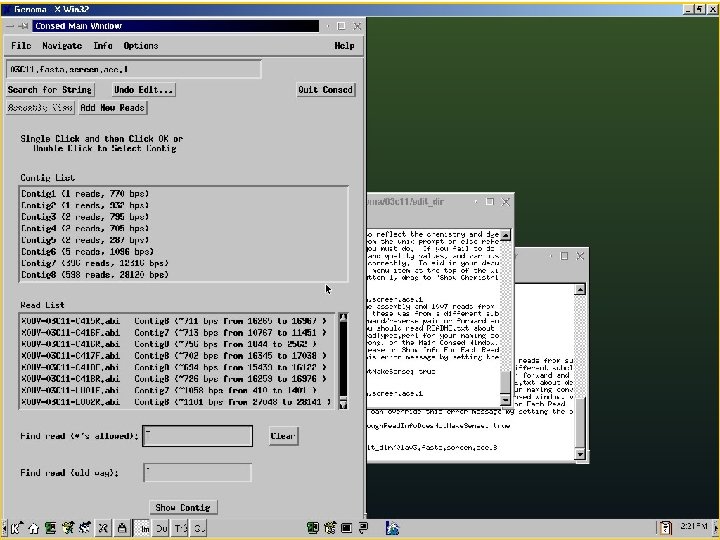
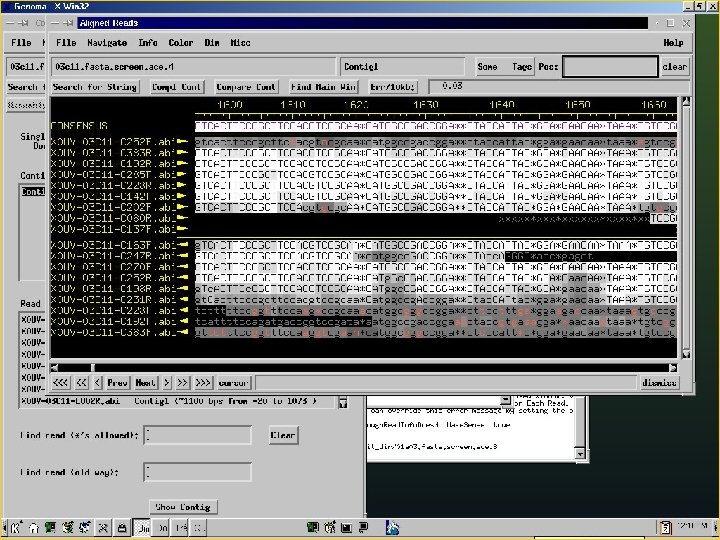
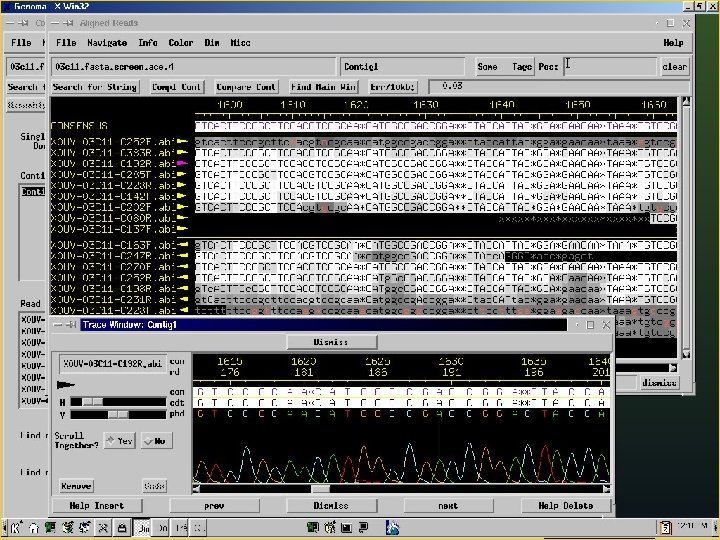

Phred/Phrap/Consed Pipeline Directories: Chromat_dir Phd_dir Edit_dir
: 47 -54")
Comparison of shotgun sequence data from Wolbachia genome Project Computer 35 (7): 47 -54

CAP 3 3 XA 6189 57 443 PHRAP 3 XA 6396 54 529 CAP 3 3 XB 12, 368 44 71 PHRAP 3 XB 13, 116 47 228 CAP 3 3 XC 10, 709 49 227 PHRAP 3 XC 11, 406 45 332 CAP 3 3 XD 11, 408 43 115 PHRAP 3 XD 11, 350 49 240 CAP 3 5 XA 10, 582 42 249 PHRAP 5 XA 18, 268 31 252 CAP 3 5 XB 26, 034 17 100 PHRAP 5 XB 33, 693 18 115 CAP 3 5 XC 20, 939 29 172 PHRAP 5 XC 20, 912 27 261 CAP 3 5 XD 14, 219 35 46 PHRAP 5 XD 14, 696 33 129

CAP 3 8 XA 71, 025 12 83 PHRAP 8 XA 71, 395 8 80 CAP 3 8 XB 53, 127 8 59 PHRAP 8 XB 53, 078 7 36 CAP 3 8 XC 52, 134 8 4 PHRAP 8 XC 76, 922 6 6 CAP 3 8 XD 72, 690 7 35 PHRAP 8 XD 102, 523 6 60 CAP 3 10 XA 91, 380 4 28 PHRAP 10 XA 91, 329 3 11 CAP 3 10 XB 167, 655 1 5 PHRAP 10 XB 138, 551 2 7 CAP 3 10 XC 106, 631 5 44 PHRAP 10 XC 77, 747 4 12 CAP 3 10 XD 79, 900 4 2 PHRAP 10 XD 79, 978 3 2
: • • • http: //genome. cs. mtu. edu/cap")
Softwares • CAP 3 (for EST): • • • http: //genome. cs. mtu. edu/cap 3. html Phrap (for large genome): http: //www. phrap. org --Similar algorithm --Insufficient documentation and support -- Always have to write scripts to parse outputs --NO PERFECT PROGRAM!!!

Questions? ? wangsj@yahoo. com
- Slides: 77