Pairwise sequence Alignment Sequence Alignment Sequence analysis is





 or more (multiple) sequences by searching for")







 Corticotropin A (pig) Oxytocin Vasopressin ala")
 H. C. Watson")





 b-lactoglobulin (P 02754) Page 42")




 family in")









 and rainbow trout (O. mykiss) 1.")
 Origin")





 considered")


 versus mouse (NP_031812) ubiquitin")













= 10 log 10 (Mab/Pb) Page 58")






 = 10")





























- Slides: 98
Pairwise sequence Alignment
Sequence Alignment • Sequence analysis is the process of making biological inferences from the known sequence of monomers in protein, DNA and RNA polymers.
Complete DNA Sequences More than 400 complete genomes have been sequenced
Evolution
Sequence alignment • Comparing DNA/protein sequences for – Similarity – Homology • Prediction of function • Construction of phylogeny • Shotgun assembly – End-space-free alignment / overlap alignment • Finding motifs
Sequence Alignment Procedure of comparing two (pairwise) or more (multiple) sequences by searching for a series of individual characters that are in the same order in the sequences GCTAGTCAGATCTGACGCTA | ||||| ||| TGGTCACATCTGCCGC
Sequence Alignment AGGCTATCACCTGACCTCCAGGCCGATGCCC TAGCTATCACGACCGCGGTCGATTTGCCCGAC -AGGCTATCACCTGACCTCCAGGCCGA--TGCCC--TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC Definition Given two strings x = x 1 x 2. . . x. M, y = y 1 y 2…y. N, an alignment is an assignment of gaps to positions 0, …, M in x, and 0, …, N in y, so as to line up each letter in one sequence with either a letter, or a gap in the other sequence
Sources of variation • Nucleotide substitution – Replication error – Chemical reaction • Insertions or deletions (indels) – Unequal crossing over – Replication slippage • Duplication – a single gene (complete gene duplication) – part of a gene (internal or partial gene duplication) • Domain duplication • Exon shuffling – part of a chromosome (partial polysomy) – an entire chromosome (aneuploidy or polysomy) – the whole genome (polyploidy)
Common mutations in DNA Substitution: A C G T T G A C G A T G A C Deletion: A C G T T G A C Insertion: A C G T T G A C G C A A T T G A C
Seq. Align. Protein 1 Protein Function Protein 2 More than 25% sequence identity ? Similar 3 D structure ? Similar function ? Similar sequences produce similar proteins
Differing rates of DNA evolution • Functional/selective constraints (particular features of coding regions, particular features in 5' untranslated regions) • Variation among different gene regions with different functions (different parts of a protein may evolve at different rates). • Within proteins, variations are observed between – surface and interior amino acids in proteins (order of magnitude difference in rates in haemoglobins) – charged and non-charged amino acids – protein domains with different functions – regions which are strongly constrained to preserve particular functions and regions which are not – different types of proteins -- those with constrained interaction surfaces and those without
Common assumptions • All nucleotide sites change independently • The substitution rate is constant over time and in different lineages • The base composition is at equilibrium • The conditional probabilities of nucleotide substitutions are the same for all sites, and do not change over time • Most of these are not true in many cases…
Pairwise alignments in the 1950 s b-corticotropin (sheep) Corticotropin A (pig) Oxytocin Vasopressin ala gly glu asp gly ala glu asp glu CYIQNCPLG CYFQNCPRG
globins: a- b- myoglobin Early example of sequence alignment: globins (1961) H. C. Watson and J. C. Kendrew, “Comparison Between the Amino-Acid Sequences of Sperm Whale Myoglobin and of Human Hæmoglobin. ” Nature 190: 670 -672, 1961.
Pairwise sequence alignment is the most fundamental operation of bioinformatics • It is used to decide if two proteins (or genes) are related structurally or functionally • It is used to identify domains or motifs that are shared between proteins • It is the basis of BLAST searching (next week) • It is used in the analysis of genomes
Pairwise alignment: protein sequences can be more informative than DNA • protein is more informative (20 vs 4 characters); many amino acids share related biophysical properties • codons are degenerate: changes in the third position often do not alter the amino acid that is specified • protein sequences offer a longer “look-back” time • DNA sequences can be translated into protein, and then used in pairwise alignments
Page 54
Pairwise alignment: protein sequences can be more informative than DNA • DNA can be translated into six potential proteins 5’ CAT CAA 5’ ATC AAC 5’ TCA ACT 5’ CATCAACTACAACTCCAAAGACACCCTTACACATCAACAAACCTACCCAC 3’ 3’ GTAGTTGATGTTGAGGTTTCTGTGGGAATGTGTAGTTGTTTGGATGGGTG 5’ 5’ GTG GGT 5’ TGG GTA 5’ GGG TAG
Pairwise alignment: protein sequences can be more informative than DNA • Many times, DNA alignments are appropriate --to confirm the identity of a c. DNA --to study noncoding regions of DNA --to study DNA polymorphisms --example: Neanderthal vs modern human DNA Query: 181 catcaactacaactccaaagacacccttacacccactaggatatcaacaaacctacccac 240 |||||| |||||||||||||||| Sbjct: 189 catcaactgcaaccccaaagccacccct-cacccactaggatatcaacaaacctacccac 247
retinol-binding protein (NP_006735) b-lactoglobulin (P 02754) Page 42
Definitions Pairwise alignment The process of lining up two or more sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology.
Definitions Homology Similarity attributed to descent from a common ancestor. Page 42
Definitions Homology Similarity attributed to descent from a common ancestor. Identity The extent to which two (nucleotide or amino acid) sequences are invariant. RBP: 26 glycodelin: 23 RVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWD- 84 + K ++ + GTW++ MA + L + A V T + + L+ W+ QTKQDLELPKLAGTWHSMAMA-TNNISLMATLKAPLRVHITSLLPTPEDNLEI V LHRWEN 81 Page 44
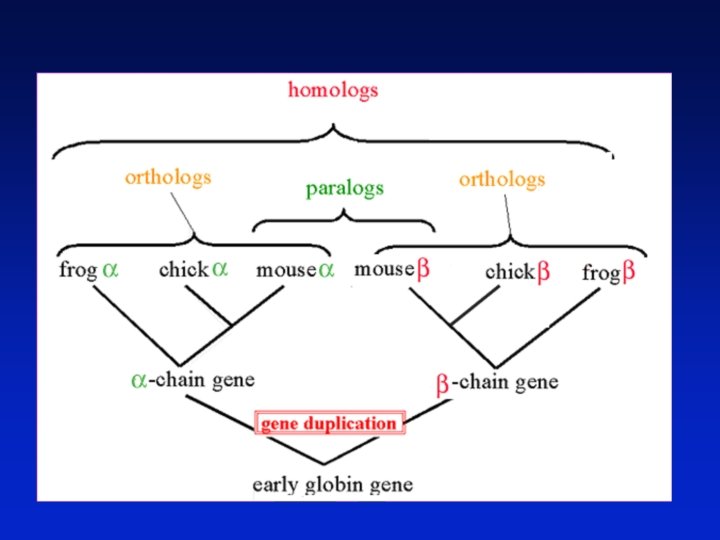
Definitions: two types of homology Orthologs Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function. Paralogs Homologous sequences within a single species that arose by gene duplication. Page 43
common carp zebrafish rainbow trout teleost Orthologs: members of a gene (protein) family in various organisms. This tree shows RBP orthologs. African clawed frog chicken human mouse rat horse pig cow rabbit 10 changes Page 43
apolipoprotein D retinol-binding protein 4 Complement component 8 Alpha-1 Microglobulin /bikunin Paralogs: members of a gene (protein) family within a species prostaglandin D 2 synthase progestagenassociated endometrial protein Odorant-binding protein 2 A neutrophil gelatinaseassociated lipocalin Lipocalin 1 10 changes Page 44
Pairwise alignment of retinol-binding protein and b-lactoglobulin 1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP. ||| |. |. . . | : . ||||. : | : 1. . . MKCLLLALALTCGAQALIVT. . QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR. LLNNWD. . VCADMVGTFTDTE 97 RBP : | | : : |. |. || |: || |. 45 ISLLDAQSAPLRV. YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV. . . QYSC 136 RBP || ||. | : . |||| |. . | 94 IPAVFKIDALNENKVL. . . . VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ. EELCLARQYRLIV 185 RBP. | | | : ||. | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI. . . . 178 lactoglobulin Page 46
Definitions Similarity The extent to which nucleotide or protein sequences are related. It is based upon identity plus conservation. Identity The extent to which two sequences are invariant. Conservation Changes at a specific position of an amino acid or (less commonly, DNA) sequence that preserve the physico-chemical properties of the original residue.
Pairwise alignment of retinol-binding protein and b-lactoglobulin 1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP. ||| |. |. . . | : . ||||. : | : 1. . . MKCLLLALALTCGAQALIVT. . QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR. LLNNWD. . VCADMVGTFTDTE 97 RBP : | | : : |. |. || |: || |. 45 ISLLDAQSAPLRV. YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV. . . QYSC 136 RBP || ||. | : . |||| |. . | 94 IPAVFKIDALNENKVL. . . . VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin Identity (bar) 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ. EELCLARQYRLIV 185 RBP. | | | : ||. | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI. . . . 178 lactoglobulin Page 46
Pairwise alignment of retinol-binding protein and b-lactoglobulin 1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP. ||| |. |. . . | : . ||||. : | : 1. . . MKCLLLALALTCGAQALIVT. . QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR. LLNNWD. . VCADMVGTFTDTE 97 RBP : | | : : |. |. || |: || |. 45 ISLLDAQSAPLRV. YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV. . . QYSC 136 RBP || ||. | : . |||| |. . | 94 IPAVFKIDALNENKVL. . . . VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin Somewhat similar (one dot) Very similar (two dots) 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ. EELCLARQYRLIV 185 RBP. | | | : ||. | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI. . . . 178 lactoglobulin Page 46
Definitions Pairwise alignment The process of lining up two or more sequences to achieve maximal levels of identity (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. Page 47
Pairwise alignment of retinol-binding protein and b-lactoglobulin 1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP. ||| |. |. . . | : . ||||. : | : 1. . . MKCLLLALALTCGAQALIVT. . QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR. LLNNWD. . VCADMVGTFTDTE 97 RBP : | | : : |. |. || |: || |. 45 ISLLDAQSAPLRV. YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV. . . QYSC 136 RBP || ||. | : . |||| |. . | 94 IPAVFKIDALNENKVL. . . . VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ. EELCLARQYRLIV 185 RBP. | | | : ||. | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI. . . . 178 lactoglobulin Internal gap Terminal gap Page 46
Gaps • Positions at which a letter is paired with a null are called gaps. • Gap scores are typically negative. • Since a single mutational event may cause the insertion or deletion of more than one residue, the presence of a gap is ascribed more significance than the length of the gap. Thus there are separate penalties for gap creation and gap extension. • In BLAST, it is rarely necessary to change gap values from the default.
Pairwise alignment of retinol-binding protein and b-lactoglobulin 1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP. ||| |. |. . . | : . ||||. : | : 1. . . MKCLLLALALTCGAQALIVT. . QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR. LLNNWD. . VCADMVGTFTDTE 97 RBP : | | : : |. |. || |: || |. 45 ISLLDAQSAPLRV. YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV. . . QYSC 136 RBP || ||. | : . |||| |. . | 94 IPAVFKIDALNENKVL. . . . VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ. EELCLARQYRLIV 185 RBP. | | | : ||. | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI. . . . 178 lactoglobulin
Pairwise alignment of retinol-binding protein from human (top) and rainbow trout (O. mykiss) 1. MKWVWALLLLA. AWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDP 48 : : || || ||. ||. . | : |||: . | ||||| 1 MLRICVALCALATCWA. . . QDCQVSNIQVMQNFDRSRYTGRWYAVAKKDP 47. . . 49 EGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTED 98 |||| ||: |||||. ||| : ||||: . ||. | || | 48 VGLFLLDNVVAQFSVDESGKMTATAHGRVIILNNWEMCANMFGTFEDTPD 97. . . 99 PAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADS 148 ||||||: ||| ||: || ||||||: : ||||| ||: ||||. . ||||| | 98 PAKFKMRYWGAASYLQTGNDDHWVIDTDYDNYAIHYSCREVDLDGTCLDG 147. . . 149 YSFVFSRDPNGLPPEAQKIVRQRQEELCLARQYRLIVHNGYCDGRSERNLL 199 |||: ||| || |||| : . . |: |. || : | |: |: 148 YSFIFSRHPTGLRPEDQKIVTDKKKEICFLGKYRRVGHTGFCESS. . . 192
Pairwise sequence alignment allows us to look back billions of years ago (BYA) Origin of life 4 Earliest fossils Origin of Eukaryote/ eukaryotes archaea 3 2 Fungi/animal Plant/animal 1 insects 0 Page 48
Multiple sequence alignment of glyceraldehyde 3 -phosphate dehydrogenases fly human plant bacterium yeast archaeon GAKKVIISAP GAKRVIISAP GAKKVVMTGP GAKKVVITAP GADKVLISAP SAD. APM. . F SKDNTPM. . F SS. TAPM. . F PKGDEPVKQL VCGVNLDAYK VMGVNHEKYD VVGVNEHTYQ VKGANFDKY. VMGVNEEKYT VYGVNHDEYD PDMKVVSNAS NSLKIISNAS PNMDIVSNAS AGQDIVSNAS SDLKIVSNAS GE. DVVSNAS CTTNCLAPLA CTTNCLAPLA CTTNSITPVA fly human plant bacterium yeast archaeon KVINDNFEIV KVIHDNFGIV KVVHEEFGIL KVINDNFGII KVINDAFGIE KVLDEEFGIN EGLMTTVHAT EGLMTTVHAI EGLMTTVHAT EGLMTTVHSL AGQLTTVHAY TATQKTVDGP TATQKTVDGP TGSQNLMDGP SGKLWRDGRG SMKDWRGGRG SHKDWRGGRT NGKP. RRRRA AAQNIIPAST ALQNIIPAST ASQNIIPSST ASGNIIPSST AAENIIPTST fly human plant bacterium yeast archaeon GAAKAVGKVI GAAKAVGKVL GAAQAATEVL PALNGKLTGM PELNGKLTGM PELQGKLTGM PELEGKLDGM AFRVPTPNVS AFRVPTANVS AFRVPTSNVS AFRVPTPNVS AFRVPTVDVS AIRVPVPNGS VVDLTVRLGK VVDLTCRLEK VVDLTVRLEK VVDLTVKLNK ITEFVVDLDD GASYDEIKAK PAKYDDIKKV GASYEDVKAA AATYEQIKAA ETTYDEIKKV DVTESDVNAA Page 49
Multiple sequence alignment of human lipocalin paralogs ~~~~~EIQDVSGTWYAMTVDREFPEMNLESVTPMTLTTL. GGNLEAKVTM LSFTLEEEDITGTWYAMVVDKDFPEDRRRKVSPVKVTALGGGNLEATFTF TKQDLELPKLAGTWHSMAMATNNISLMATLKAPLRVHITSEDNLEIVLHR VQENFDVNKYLGRWYEIEKIPTTFENGRCIQANYSLMENGNQELRADGTV VKENFDKARFSGTWYAMAKDPEGLFLQDNIVAEFSVDETGNWDVCADGTF LQQNFQDNQFQGKWYVVGLAGNAI. LREDKDPQKMYATIDKSYNVTSVLF VQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVDGGLNLTSTFL VQENFNISRIYGKWYNLAIGSTCPWMDRMTVSTLVLGEGEAEISMTSTRW PKANFDAQQFAGTWLLVAVGSACRFLQRAEATTLHVAPQGSTFRKLD. . . lipocalin 1 odorant-binding protein 2 a progestagen-assoc. endo. apolipoprotein D retinol-binding protein neutrophil gelatinase-ass. prostaglandin D 2 synthase alpha-1 -microglobulin complement component 8 Page 49
General approach to pairwise alignment • Choose two sequences • Select an algorithm that generates a score • Allow gaps (insertions, deletions) • Score reflects degree of similarity • Alignments can be global or local • Estimate probability that the alignment occurred by chance
Calculation of an alignment score
Where we’re heading in the next 10 minutes: creating a set of “scoring matrices” that let us assign scores for each aligned amino acid in a pairwise alignment. What should the score be when a serine matches a serine, or a threonine, or a valine? Can we devise “lenient” scoring systems to help us align distantly related proteins, and more conservative scoring systems to align closely related proteins?
lys found at 58% of arg sites Emile Zuckerkandl and Linus Pauling (1965) considered substitution frequencies in 18 globins (myoglobins and hemoglobins from human to lamprey). Black: identity Gray: very conservative substitutions (>40% occurrence) White: fairly conservative substitutions (>21% occurrence) Red: no substitutions observed Page 80
Page 80
Dayhoff’s 34 protein superfamilies Accepted point mutations Protein Ig kappa chain Kappa casein Lactalbumin Hemoglobin a Myoglobin Insulin Histone H 4 Ubiquitin From 1978 PAMs per 100 million years 37 33 27 12 8. 9 400 fold 4. 4 0. 10 0. 00 Page 50
Pairwise alignment of human (NP_005203) versus mouse (NP_031812) ubiquitin
Multiple sequence alignment of glyceraldehyde 3 -phosphate dehydrogenases fly human plant bacterium yeast archaeon GAKKVIISAP GAKRVIISAP GAKKVVMTGP GAKKVVITAP GADKVLISAP SAD. APM. . F SKDNTPM. . F SS. TAPM. . F PKGDEPVKQL VCGVNLDAYK VMGVNHEKYD VVGVNEHTYQ VKGANFDKY. VMGVNEEKYT VYGVNHDEYD PDMKVVSNAS NSLKIISNAS PNMDIVSNAS AGQDIVSNAS SDLKIVSNAS GE. DVVSNAS CTTNCLAPLA CTTNCLAPLA CTTNSITPVA fly human plant bacterium yeast archaeon KVINDNFEIV KVIHDNFGIV KVVHEEFGIL KVINDNFGII KVINDAFGIE KVLDEEFGIN EGLMTTVHAT EGLMTTVHAI EGLMTTVHAT EGLMTTVHSL AGQLTTVHAY TATQKTVDGP TATQKTVDGP TGSQNLMDGP SGKLWRDGRG SMKDWRGGRG SHKDWRGGRT NGKP. RRRRA AAQNIIPAST ALQNIIPAST ASQNIIPSST ASGNIIPSST AAENIIPTST fly human plant bacterium yeast archaeon GAAKAVGKVI GAAKAVGKVL GAAQAATEVL PALNGKLTGM PELNGKLTGM PELQGKLTGM PELEGKLDGM AFRVPTPNVS AFRVPTANVS AFRVPTSNVS AFRVPTPNVS AFRVPTVDVS AIRVPVPNGS VVDLTVRLGK VVDLTCRLEK VVDLTVRLEK VVDLTVKLNK ITEFVVDLDD GASYDEIKAK PAKYDDIKKV GASYEDVKAA AATYEQIKAA ETTYDEIKKV DVTESDVNAA
Dayhoff’s numbers of “accepted point mutations”: what amino acid substitutions occur in proteins? From closely related protein sequences (at least 85% identity) Numbers of APM, multiplied by 10, in 1572 cases of amino acid substitutions from closely related sequences
The relative mutability of amino acids Asn Ser Asp Glu Ala Thr Ile Met Gln Val 134 120 106 102 100 97 96 94 93 74 His Arg Lys Pro Gly Tyr Phe Leu Cys Trp 66 65 56 56 49 41 41 40 20 18 Describes how often each amino acid is likely to change over a short evolutionary period Page 53
Normalized frequencies of amino acids Gly Ala Leu Lys Ser Val Thr Pro Glu Asp 8. 9% 8. 7% 8. 5% 8. 1% 7. 0% 6. 5% 5. 8% 5. 1% 5. 0% 4. 7% Arg Asn Phe Gln Ile His Cys Tyr Met Trp 4. 1% 4. 0% 3. 8% 3. 7% 3. 4% 3. 3% 3. 0% 1. 5% 1. 0% blue=6 codons; red=1 codon Page 53
Page 54
Dayhoff’s PAM 1 mutation probability matrix Original amino acid Replaced amino acid There is 98. 67%chance that A will be replaced by A over an evolutionary distance of 1 PAM Each element shows the probability that an original amino acid j (columns)will be replaced byanother amino acid i (rows) for 1% sequence divergence
Dayhoff’s PAM 1 mutation probability matrix Each element of the matrix shows the probability that an original amino acid (top) will be replaced by another amino acid (side)
Substitution Matrix A substitution matrix contains values proportional to the probability that amino acid i mutates into amino acid j for all pairs of amino acids. Substitution matrices are constructed by assembling a large and diverse sample of verified pairwise alignments (or multiple sequence alignments) of amino acids. Substitution matrices should reflect the true probabilities of mutations occurring through a period of evolution. The two major types of substitution matrices are PAM and BLOSUM.
PAM matrices: Point-accepted mutations PAM matrices are based on global alignments of closely related proteins. The PAM 1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. Other PAM matrices are extrapolated from PAM 1. All the PAM data come from closely related proteins (>85% amino acid identity)
Dayhoff’s PAM 1 mutation probability matrix Page 55
Dayhoff’s PAM 0 mutation probability matrix: the rules for extremely slowly evolving proteins Top: original amino acid Side: replacement amino acid Page 56
Dayhoff’s PAM 2000 mutation probability matrix: the rules for very distantly related proteins PAM A R N D C Q E G Ala Arg Asn Asp Cys Gln Glu Gly A 8. 7% 8. 7% R 4. 1% 4. 1% N 4. 0% 4. 0% D 4. 7% 4. 7% C 3. 3% 3. 3% Q 3. 8% 3. 8% E 5. 0% 5. 0% G 8. 9% 8. 9% PAM 1 matrix is multiplied 2000 times by itself Top: original amino acid Side: replacement amino acid
PAM 250 mutation probability matrix Top: original amino acid Side: replacement amino acid Page 57
PAM 250 log odds scoring matrix S(a, b)= 10 log 10 (Mab/Pb) Page 58
Why do we go from a mutation probability matrix to a log odds matrix? • We want a scoring matrix so that when we do a pairwise alignment (or a BLAST search) we know what score to assign to two aligned amino acid residues. • Logarithms are easier to use for a scoring system. They allow us to sum the scores of aligned residues (rather than having to multiply them). Page 57
How do we go from a mutation probability matrix to a log odds matrix? • The cells in a log odds matrix consist of an “odds ratio”: the probability that an alignment is authentic the probability that the alignment was random The score S for an alignment of residues a, b is given by: S(a, b) = 10 log 10 (Mab/pb) As an example, for tryptophan, Normalized frequency of W is 0. 01 S(a, tryptophan) = 10 log 10 (0. 55/0. 010) = 17. 4
What do the numbers mean in a log odds matrix? S(a, tryptophan) = 10 log 10 (0. 55/0. 010) = 17. 4 A score of +17 for tryptophan means that this alignment is 50 times more likely than a chance alignment of two Trp residues. S(a, b) = 17 Probability of replacement (Mab/pb) = x Then 17 = 10 log 10 x 1. 7 = log 10 x 101. 7 = x = 50 Page 58
What do the numbers mean in a log odds matrix? A score of +2 indicates that the amino acid replacement occurs 1. 6 times as frequently as expected by chance. A score of 0 is neutral. A score of – 10 indicates that the correspondence of two amino acids in an alignment that accurately represents homology (evolutionary descent) is one tenth as frequent as the chance alignment of these amino acids. Page 58
PAM 250 log odds scoring matrix Page 58
PAM 10 log odds scoring matrix Page 59
Rat versus mouse RBP Rat versus bacterial lipocalin
Comparing two proteins with a PAM 1 matrix gives completely different results than PAM 250! Consider two distantly related proteins. A PAM 40 matrix is not forgiving of mismatches, and penalizes them severely. Using this matrix you can find almost no match. hsrbp, 136 CRLLNLDGTC btlact, 3 CLLLALALTC * ** A PAM 250 matrix is very tolerant of mismatches. 24. 7% identity in 81 residues overlap; Score: 77. 0; Gap frequency: 3. 7% hsrbp, 26 RVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDV btlact, 21 QTMKGLDIQKVAGTWYSLAMAASD-ISLLDAQSAPLRVYVEELKPTPEGDLEILLQKWEN * **** * * ** * hsrbp, 86 --CADMVGTFTDTEDPAKFKM btlact, 80 GECAQKKIIAEKTKIPAVFKI ** ** Page 60
BLOSUM Matrices BLOSUM matrices are based on local alignments. BLOSUM stands for blocks substitution matrix. BLOSUM 62 is a matrix calculated from comparisons of sequences with no less than 62% divergence. Page 60
62 co l e ps la Percent amino acid identity BLOSUM Matrices 100 30 BLOSUM 62
100 e lla 100 30 e ps 30 62 lla 62 co l la ps co Percent amino acid identity 100 ps e BLOSUM Matrices BLOSUM 80 BLOSUM 62 30 BLOSUM 30
BLOSUM Matrices All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins. The BLOCKS database contains thousands of groups of multiple sequence alignments. BLOSUM 62 is the default matrix in BLAST 2. 0. Though it is tailored for comparisons of moderately distant proteins, it performs well in detecting closer relationships. A search for distant relatives may be more sensitive with a different matrix. Page 60
BLOSUM Scoring Matrices • In the Dayhoff model, the scoring values are derived from protein sequences with at least 85% identity • Alignments are, however, most often performed on sequences of less similarity, and the scoring matrices for use in these cases are calculated from the 1 PAM matrix • Henikoff and Henikoff (1992) have therefore developed scoring matrices based on known alignments of more diverse sequences
BLOSUM Scoring Matrices • They take a group of related proteins and produce a set of blocks representing this group, where a block is defined as an ungapped region of aligned amino acids • An example of two blocks is KIFIMK NLFKTR KIFKTK KLFESR KIFKGR GDEVK GDSKK GDPKA GDAER GDAAK
• The Henikoffs used over 2000 blocks in order to derive their scoring matrices • For each column in each block they counted the number of occurrences of each pair of amino acids, when all pairs of segments were used • Then the frequency distribution of all 210 different pairs of amino acids were found • A block of length w from an alignment of m sequences makes (wm(m-1))/2 pairs of amino acids
We define • hab as the number of occurrences of the amino acid pair (ab) (note that hab=hba) • T as the total number of pairs in the alignment where ≥ is interpreted as a total ordering over the amino acids • fab=hab/T (the frequency of observed pairs)
Developing Scoring Matrices for Different Evolutionary Distances • The procedure for developing a BLOSUM X matrix 1. Collect a set of multiple alignments 2. Find the blocks 3. Group the segments with an X% identity 4. Count the occurrences of all pairs of amino acids 5. Develop the matrix, as explained before • BLOSUM-62 is often used as the standard for ungapped alignments • For gapped alignments, BLOSUM-50 is more often used
Blosum 62 scoring matrix Page 61
By use of relative entropy, it can be found that PAM 250 corresponds to BLOSUM-45 and PAM 160 corresponds to BLOSUM-62, and PAM 120 corresponds to BLOSUM-80 Rat versus mouse RBP Rat versus bacterial lipocalin Page 61
Major Differences between PAM and BLOSUM
PAM matrices: Point-accepted mutations PAM matrices are based on global alignments of closely related proteins. The PAM 1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. Other PAM matrices are extrapolated from PAM 1. All the PAM data come from closely related proteins (>85% amino acid identity)
Percent identity Two randomly diverging protein sequences change in a negatively exponential fashion “twilight zone” Evolutionary distance in PAMs Page 62
Percent identity At PAM 1, two proteins are 99% identical At PAM 10. 7, there are 10 differences per 100 residues At PAM 80, there are 50 differences per 100 residues At PAM 250, there are 80 differences per 100 residues “twilight zone” Differences per 100 residues PAM 250 PAM matrices reflect different degrees of divergence Page 62
PAM: “Accepted point mutation” • Two proteins with 50% identity may have 80 changes per 100 residues. (Why? Because any residue can be subject to back mutations. ) • Proteins with 20% to 25% identity are in the “twilight zone” and may be statistically significantly related. • PAM or “accepted point mutation” refers to the “hits” or matches between two sequences (Dayhoff & Eck, 1968) Page 62
Ancestral sequence ACCCTAC A C C C --> G T --> A A --> C --> T C no change single substitution multiple substitutions coincidental substitutions parallel substitutions convergent substitutions back substitution Sequence 1 A C --> A --> T C --> A T --> A A --> T C --> T --> C Sequence 2 Li (1997) p. 70
Percent identity between two proteins: What percent is significant? 100% 80% 65% 30% 23% 19%
An alignment scoring system is required to evaluate how good an alignment is • positive and negative values assigned • gap creation and extension penalties • positive score for identities • some partial positive score for conservative substitutions • global versus local alignment • use of a substitution matrix