Caso clnico Prpura Trombocitopnica Idiopatica ESCS Pediatria HRAS

Caso clínico: Púrpura Trombocitopênica Idiopatica ESCS – Pediatria - HRAS Frederico Naves Godoi Coordenação: Luciana Sugai www. paulomargotto. com. br - 5/8/2009

l Identificação: LGC, 3 anos e 4 meses, natural de Brasília/DF, procedente do Guara/DF. l QP: manchas no corpo há 6 dias. l HDA: paciente iniciou , subitamente, há cerca de 6 dias com manchas “arroxeadas” (equimoses) em membros inferiores que evoluíram para tronco e membro superiores. Variavam de tamanho e não estavam associadas a dor ou a trauma local. Observado também petéquias em face. Nega febre, vômito e alteração do hábito intestinal. Nega sangramentos. Aceitava dieta e se mantinha ativo.

l Antecedente: l l l Há 2 meses criança teve otite média aguda, tratada com amoxacilina. Há cerca de 25 dias iniciou febre e amigdalite, tratada com cefalexina, inicialmente, e substituída por amoxacilina + clavulanato com remissão do quadro. Nega internações prévias e hemo-transfusão. Nega alergia. Nega cirurgia Nascida de parto cesária, a termo e sem complicações. Gestação normal com 7 consultas de pré-natal. Vacinação completa. Pais hígidos. Nega co-sanguinidade. Mora em casa de alvenaria com rede de esgoto, água encanada, energia elétrica e coleta de lixo. Nega animais.

Exame físico: l Tax: 36, 4°C; FC: 98 bpm; FR: 33 ipm. l Ectoscopia: BEG, afebril, hidratado, eupnéico, acianótico, anictérico, hipocorado(+/4+), ativo. l Não evidenciado cadeia ganglionar aumentada ou dolorosa. l Orofaringe sem alteração. l AR: tórax simétrico, MVF sem ruídos l ACV: ictus visível em linha hemiclavicular esquerda no 5° espaço intercostas, sem frêmitos à palpação; BNF, RCR em 2 T sem sopro.

l ABD: simétrico, normotense, RHA+, timpânico, indolor, sem massas ou VMG l Ext: perfundida e sem edema. l SNC: sem sinais meníngeos; sensibilidade e movimentação preservada; orientado e ativo; reflexos sem alteração. l Pele: mancha purpúrica (equimose) em membro e tronco anterior e posterior de tamanho variável, planas e indolores; petéquias em face.

Exames laboratoriais: Hc: Hb: 11. 2 Ht: 34. 1 l Leuc: 11. 4 (55 0 20 0 0) l Plaquetas: 18. 000 l VHS: 3 l TGP: 18 TGP: 12 l Albumina: 4. 6 l TS: TAP: l

Hipótese diagnóstica ?
")
Hipótese diagnóstica Púrpura trobocitopência idiopática (PTI)

Hemostasia: l Hemostasia primária: estacar o sangramento pela formação do trombo plaquetário l Hemostasia secundária: evita o ressangramento pela formação de uma rede de fibrina que consolida o trombo (coágulo).

hemostasia l Primária: l Secundária: l Adesão/ativação/agregação plaquetaria Predomina pele/mucosa Gengivorragia, epistaxe, menorragia, petéquias e equimoses Persistência do sangramento, de pq qti, após cortes superficiais Contagem de plaquetas, índices plaquetários e TS. l Proteínas(fatores de coagulação) que serão ativadas formando a rede de fibrina Predomina órgãos/tecidos internos Hemartrose, hematoma dissecante profundo ou muscular Sangramento recidivante, em grande monta e difícil controle TC, PTTa, TAP e INR l l l l

Distúrbios plaquetários: l Pseudotrombocitopenia: falsa baixa concentração pelo aparelho de automação→fazer contagem manual l Destruição acelerada: causa mais comum. Aumento do consumo ou destruição de plaquetas estimula a trombopoiese, aumento do número, tamanho e taxa de maturação dos precursores megacariocíticos l l Mecanismos imunes: PTI idiopática, relacionada ao LES ou ao HIV, induzida por drogas e por algumas infecções. Mecanismos não-imunes: PTT, SHU e a CIVD

l Diminuição da produção pela medula óssea: mielodisplasias, anemia aplásica e leucemia. A biópsia e o aspirado nos fornecem o diagnóstico correto na maioria dos casos. l Trombopoiese ineficaz: carência de vitamina B 12 ou ácido fólico (análogo à eritropoiese ineficaz). l Distribuição anormal: hiperesplenismo (hipertensão portal ou doenças infiltrativas). l Trombocitopenia dilucional: observada durante a transfusão maciça de concentrado de hemácias (+ de 8 unidades de sangue em 24 horas). Não é necessária a reposição rotineira de plaquetas nesses casos.

PTI

PTI l Destruição de plaquetas de forma prematura na circulação l Auto-anticorpos ou imunocomplexos na superfície da membrana (opsonizadas por Ig. G) l Anticorpos direcionados para sítios da glicoproteína IIb/IIIa l Sistema reticuloendotelial do baço e fígado. l Vida-média de algumas horas (normal de 7 a 10 dias) l Nenhuma doença ou condição subjacente é identificada
: l l l l Trombocitopenia <6 meses")
Formas de apresentação l Forma infantil (aguda): l l l l Trombocitopenia <6 meses Crianças e adultos jovens (pico 2 -6 anos) Em 75% dos casos→após infecção viral respiratória ou exantemática ou à vacinação (menos comum) Plaquetopenia importante (< 20. 000) Pouca frequência de sangramento grave Resolução espontânea em 90% dos casos (4 a 6 sem. ); 10% se cronifica Diagnóstico diferencial: púrpura de Henoch-Schonlein, leucemias agudas, meningococcemia e estafilococcemia, SHU
: l Relação mulher/homem de 3: 1")
Formas de apresentação l Forma do adulto (crônica): l Relação mulher/homem de 3: 1 na faixa de 20 -40 anos l Início insidioso; história de sintomas hemorrágicos de gravidade variável l Geralmente sem antecedentes de infecção l Marca: curso flutuante de sangramento, por alguns dias a semanas, de caráter intermitente ou cíclico l Remissão espontânea rara (10%)

Manifestações clínicas: l l l l Gravidade da hemorragia variável, de acordo com a contagem de plaquetas < 10. 000 – ameaça à vida (hemorragia cerebral) Petéquias e equimoses em região dorsal e coxa e em áreas de estase vascular Sangramentos intensos são raros Gengivorragia, bolhas hemorrágicas e epistaxe responsivo a tamponamento Sangramento menstrual excessivo Sangramento geniturinário e intestinal (melena) Sangramento em SNC (1% dos casos): hemorragia subaraquinóide e peq. sangramento na retina e subconjutival
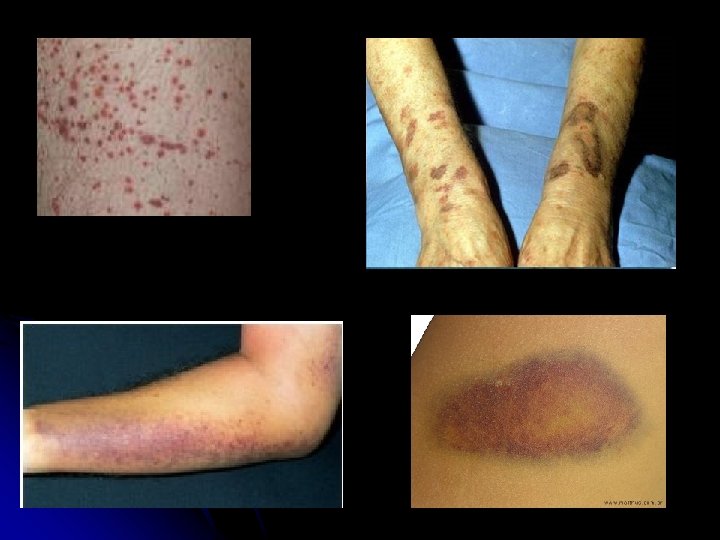
 e: l")
diagnóstico l Exclusão em paciente com trombocitopenia (geralmente inferior a 50. 000) e: l l l Ausência de esquizócitos, blastos ou outra etiologia trombocitopênica Ausência de causas secundárias (HIV, LES, drogas, LLC) Aspirado de medula óssea: afastar outras doenças hematológicas como leucemia e mielodisplasias Anticorpo antiplaquetário - ↓ acurácia Laboratório: -MPV>12 f. L -PDW elevado -leucocitos e hemoglobina normais, embora possa revelar anemia e leucocitose como resposta ao sangramento agudo
 l")
tratamento l Plaq. > que 30. 000→orientar evitar traumas e drogas antiplaquetária (AAS) l Plaq. < que 30. 000 com sangramento→prednisona 2 mg/kg/dia, por 2 -4 sem. , seguida de redução também em 4 -6 sem. l Alguns autores recomendam tratar criança assintomática com plaq. < 20. 000 l Casos crônico: tratar se <20. 000 ou se <50. 000 com sangramento. O restante pode ser acompanhado l Remissão completa: plaquetas > ou igual a 100. 000 mantida por 6 meses (10 -30% dos casos)

l Corticóide: l l l tratamento Reduz a fenestração endotelial Reduz os receptores Fcγ dos macrófagos esplênicos Inibi a produção de anticorpos pelo baço e medula óssea l Resposta objetiva em 70% dos caso, ou seja, plaquetometria > 50. 000 após 2 -4 sem. de uso l 90% voltam a fazer plaquetopenia após a redução da dose. Se > que 20. 000 -30. 000 e sem sangramento, podem ser apenas observados. l É aceitável à longo prazo: 10 mg em dias alternados

tratamento l Imunoglobulina: ocupa os receptores Fcγ dos macrófagos l Apesar de onerosa, é a de maior eficácia a curto prazo l Alternativa à PTI refratária l Resposta em 85%, mas mantida apenas por um mês l l Droga de escolha em caso de dúvida do diagnóstico, sem a possibilidade de fazer o aspirado de medula óssea

tratamento l Esplenectomia eletiva: l l Esplenectomia de urgência: l l Pacientes que recaem com a diminuição da prednisona <20. 000 -30. 000 por mais de 3 meses Trombocitopenia grave (<10. 000) que não respondem à terapia esteróide em 6 semanas Não resposta em 20 -50% dos casos – tecido esplênico acessório

tratamento l Transfusão de plaquetas na PTI: l. A princípio não está indicada l Estudos recentes mostram resposta satisfatória na abordagem da plaquetopenia grave sintomática na PTI

tratamento l PTI secundária: l Tratar causa básica l Suspender droga, se for o caso l No HIV-SIDA tratar com zidovudinha (AZT), evitando os corticóides

Púrpura de Henoch-Schonlein l Vasculite sistêmica de pequenos vasos, sendo a mais comum na infância l Predomina no sexo masculino e idade média de início aos 5 anos (3 -20) l Etiologia desconhecida 50% precedida por infecção do trato respiratório superior. l Drogas desencadeantes (penicilina, eritromicina, quinidina) l l Mecanismos imunológico: hiperprodução de Ig. A e deposição tecidual de imunocomplexos
")
Manifestações clínicas l l Púrpura palpável→principalmente em nádegas e membros inferiores (100% dos casos) Poliartralgia e artrite: mais comum em joelho e tornozelo Dor abdominal, náuses, vômitos e diarréia com muco e sangue decorrente da vasculite mucosa Lesão renal (40 -50%) l l Geralmente branda (hematúria) e assintomática Alterações nefríticas discretas até GNRP Deposição de Ig. A no mesângio→Doença de Berger Autolimitada, normalmente de 4 -6 semanas, sendo comum haver 2 ou mais recidivas, especialmente no primeiro ano após o surto inicial

diagnóstico l Essencialmente clínico l Tétrade de púrpura, artralgia/artrite, dor abdominal e hamatúria l Anemia e leucocitose leves l Plaqueta e testes de coagulação normais l Complemento normal l Biópsia das lesões cutâneas: típicas vasculite leucocitoclásticas (debris de núcleos de neutrófilos)

Tratamento l Maioria dos caso necessitam apenas de repouso e analgésicos l Se dor abdominal intensa, sangramento digestivo ou nefrite grave→prednisona 1 -2 mg/kg/dia por 2 semanas, reduzindo -se gradualmente a dose em curto período de tempo l Já forem descritos usos de azatioprina, ciclofosfamida, ciclosporina, gamaglobulina e plasmaferese com resultados variados.
 l l Causa mais comum de IRA intrínseca em menores de")
Síndrome hemolítico-urêmica (SHU) l l Causa mais comum de IRA intrínseca em menores de 4 anos Maioria das vezes, 7 -10 dias após gastroenterite com diarréia sanguinolenta, causada pelo E. coli Incidência maior em países subdesenv. de clima quente Etiologia desconhecida l lesão endotelial glomerular→resposta com hiperatividade plaquetária e do sist. de coagulação local Fv. WB aumentado com efeito ativador de plaquetas Acúmulo nas alças capilares glomerulares levando a obstrução glomerular

Diagnóstico/manifestação clínica l Tríade clínica Anemia hemolítica microangiopática l Plaquetopenia l Insuficiência renal aguda oligúrica l l Histopatológico renal: depósito subendotelial e mesangial l Esfregaço sangue periférico: esquizócitos (fagmentos de hemácias) l Leucocitose (em torno de 20. 000) l Aumento das escórias nitrogenadas l EAS: hematúria microscópia, proteinúria e cilindros granulares

Tratamento l l l Não tem tratamento específico Diálise peritoneal é suficiente para tratar uremia e distúrbios hidroeletrolíticos Auto-limitada na maioria das vezes Alguns autores indicam heparina em baixa dose para evitar a formação de fibrina no glomérulo e também a plasmaferese, porém sem benefícios documentados 10% evoluem para necrose tubular aguda e alguns para rins em fase terminal, necessitando de diálise para o resto da vida.

obrigado
- Slides: 33