LE TALASSEMIE TALASSEMIE ANEMIA MEDITERRANEA Difetti genetici della

 Difetti genetici della sintesi di una o più catene globiniche normali")























 Individui con alpha Thalassemia grave e Hb Barts soffrono di")









 COMPOSIZIONE Hb. A (96 -97) 2 2")
 1")


 2.")


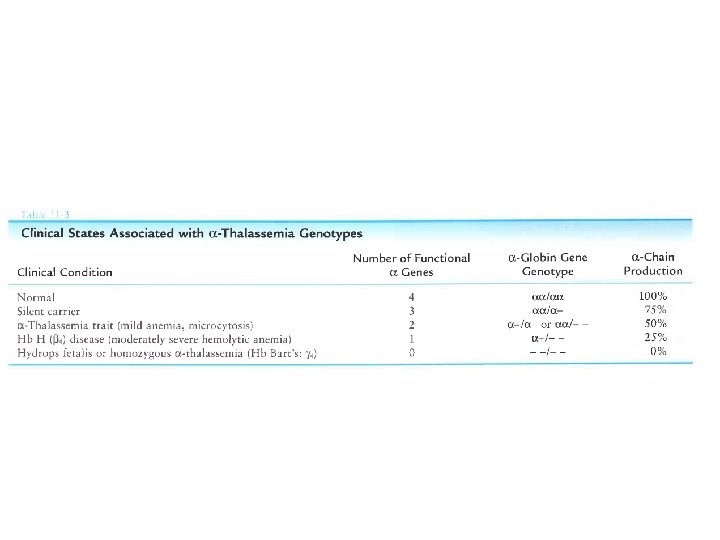
 GENOTIPO HB FENOTIPO")







, ipocromica, microcitica,")







: range ampio di gravità da quello di")














- Slides: 86
LE TALASSEMIE
TALASSEMIE (ANEMIA MEDITERRANEA) Difetti genetici della sintesi di una o più catene globiniche normali Inadeguata produzione di emoglobina Anemia ipocromica-microcitica Sintesi bilanciata di catene globiniche, precipitazione di tetrameri instabili Eritropoiesi inefficace ed emolisi
EMOGLOBINOPATIE • Difetti genetici della sintesi emoglobinica con produzione di catene peptidiche anomale nella sequenza aminoacidica (sostituzione, perdita o aggiunta di aminoacidi) • Differenti genotipi e fenotipi: Hb. S (anemia falciforme), Hb. C, Hb. H, metaemoglobinopatie, Hb con ridotta o aumentata affinità per O 2
Talassemie derivano dal greco “mare” per essere state descritte per la prima volta in pazienti di origine mediterranea. Elevata frequenza Talassemie per effetto protettivo stato eterozigote contro malaria Mutazioni comportano riduzione livelli catene a or Inadequata produzione di Hb determina hypochromia e microcytosis Catena prodotta normalmente diventa problema in quanto è in eccesso Accumulo non bilanciato provoca eritropoiesi inefficace e anemia emolitica
EMOGLOBINA
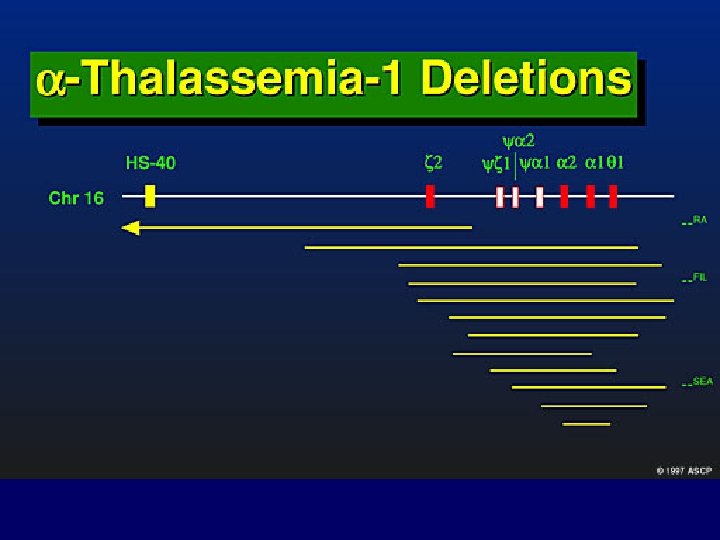
Alpha-Thalassemias • più comuni delle beta thalassemie • difetti delle catene alfa alterano formazione delle emoglobine fetali e adulte, malattia intrauterina e fetale • probabile meccanismo delezione gene alfa: per allineamento non corretto e appaiamento tra geni a 1 e a 2 seguito da ricombinazione
TALASSEMIE • Diffuse prevalentemente nel Sud-Est asiatico • Quadri clinico-ematologici variabili a seconda del grado di riduzione nella produzione delle catene • Se mancano 1 -2 geni : – quadro clinico sovrapponibile a talassemia minor. – Hb Bart ( 4)alla nascita 5 -10%, Hb normale in età adulta (diagnosi di esclusione) • Se mancano 3 -4 geni : – idrope fetale (80% Hb Bart): incompatibile con la vita – malattia da Hb. H ( 4). Severa anemia ipocromica, microcitica, con componente emolitica, splenomegalia.
DIFFERENTI TIPI DI TALASSEMIE
Malattia da Hb. H: reticolociti e eritrociti con aggregati di catene Talassemia : Ipocromia, microcitosi, cellule bersaglio e poichilocitosi
Hp = aptoglobina
il più comune
triplicated locus observed occasionally - selective disadvantage ?
meno comune
inclusioni da Hb. H più numerose
Regione regolatoria Emoglobina Z se presente gene z increased z-globin expression
Base genetica di thalassemia: • sopratutto delezioni chr. 16 2 1 2 2 2 1 1 Neither release O 2 hemoglobin normale Portatore silente Lieve anemia microcitica clinicamente benigna Anemia grave clinicamente Hb Barts ( 4) severa Hb H ( 4) hydrops fetalis Tratto talassemico
Hydrope fetalis (homozygous -Thalassemia) Individui con alpha Thalassemia grave e Hb Barts soffrono di grave ipossia intrauterine e nascono con massiccio accumulo di liquidi
Pathogenesis of Hydrops Severe anemia Hepatic extramedullary hematopoeisis Decreased prdtn of plasma proteins Decreased plasma COP
Pathogenesis of Hydrops Congestive heart failure Increased central venous pressure Increased capillary hydrostatic pressure
Pathogenesis of Hydrops Severe tissue hypoxia Endothelial cell damage Capillary leak of fluid & protein
Decreased Increased Pressione osm coll Pressione idrost vasc Increased fluid efflux from intravascular space Capillary leak
The Fetal Microcirculation Fetal Sheep Adult Sheep 1 l NS bolus IV 30% intravascular at 30 min 1 l NS bolus IV 6% intravascular at 30 min Aumento nel feto della permeabilità capillare e della capacità assorbimento spazio interstiziale
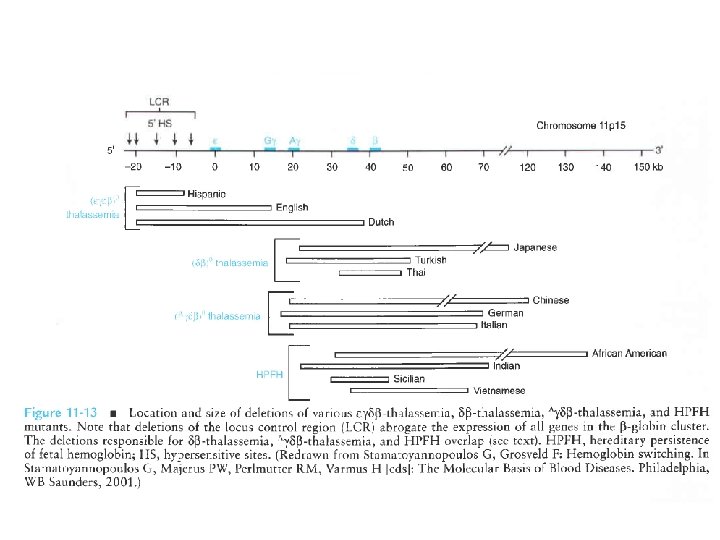
BETA-TALASSEMIA, COMPLEX TALASSEMIA, AND HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN
LE BETA-TALASSEMIE
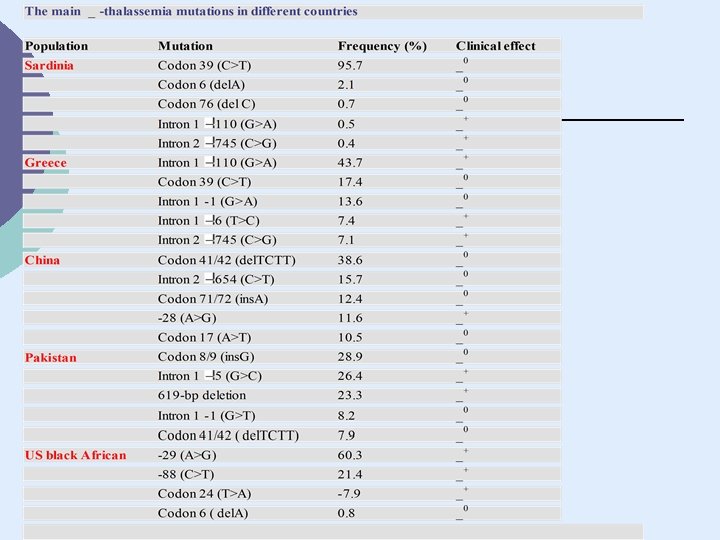
DISTRIBUZIONE GEOGRAFICA DELLE TALASSEMIE E DELLE EMOGLOBINOPATIE
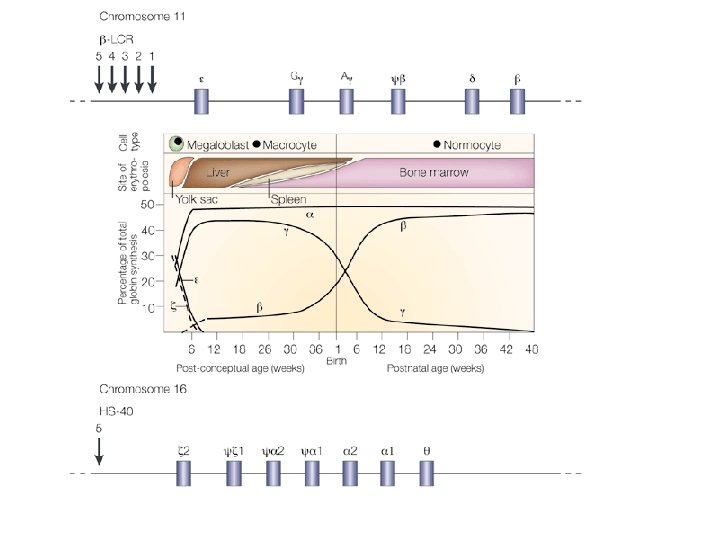
ONTOGENESI DELL’EMOGLOBINA E SEDI DI PRODUZIONE Cromos. 16 Cromos. 11
TIPI DI EMOGLOBINA NELL’ADULTO NORMALE EMOGLOBINA (%) COMPOSIZIONE Hb. A (96 -97) 2 2 Hb. A 2(2 -3) 2 2 Hb. F(0 -1) 2 2
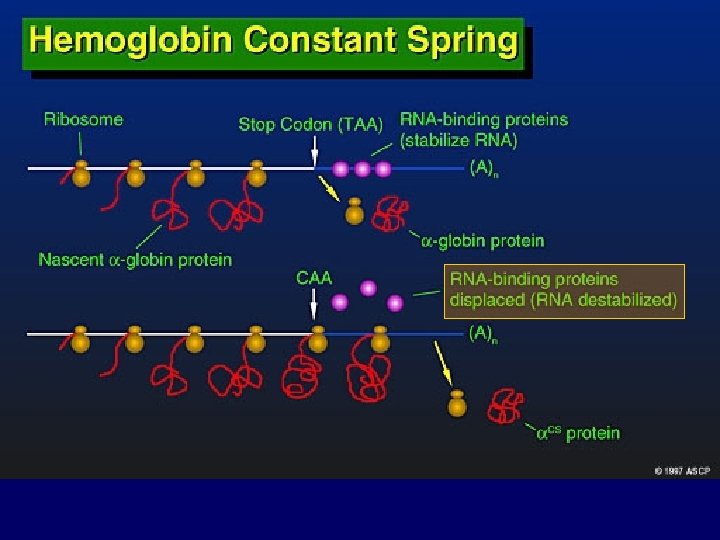
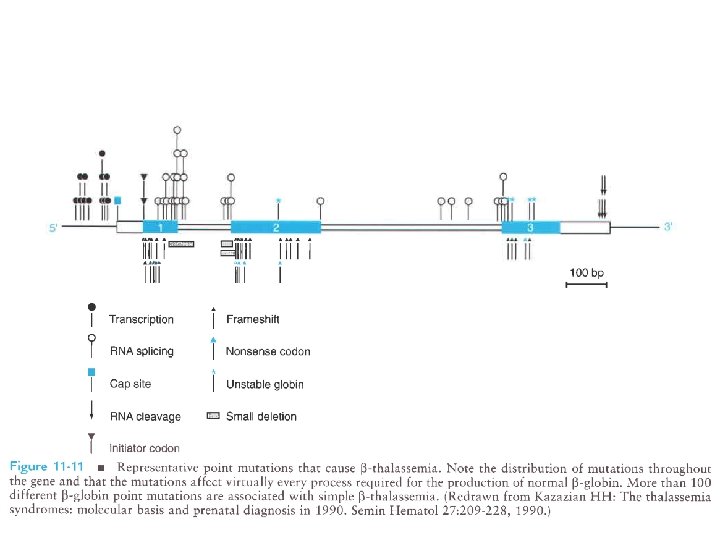
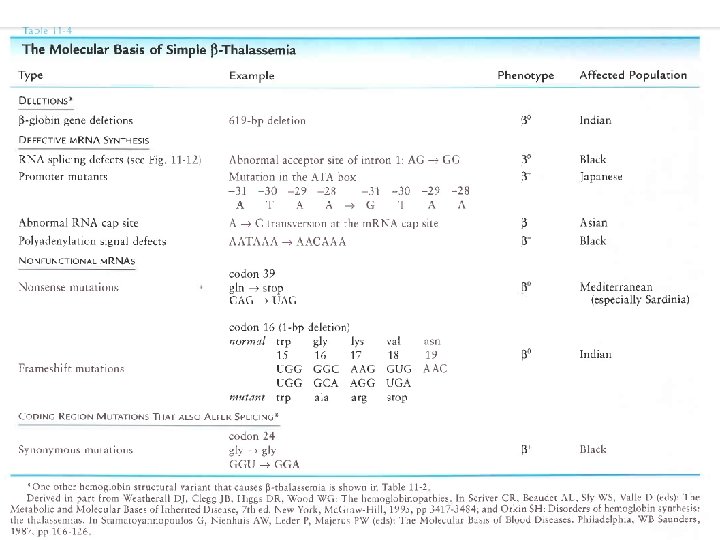
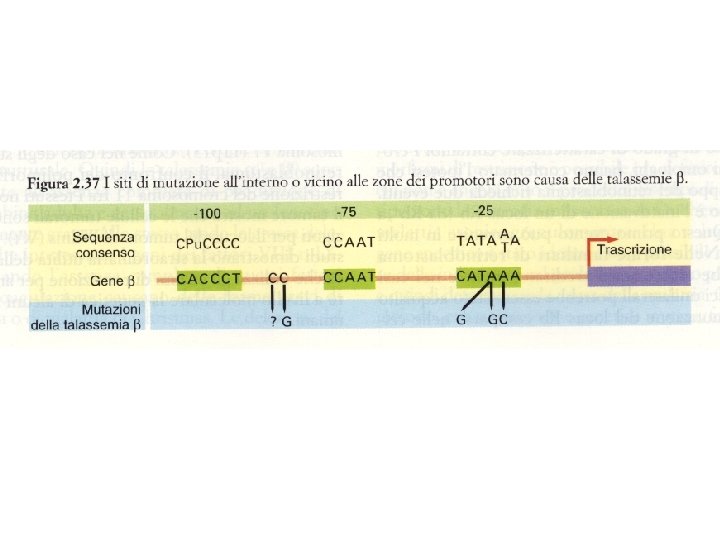
MECCANISMI MOLECOLARI CHE PRODUCONO LE TALASSEMIE 1. Delezione genica (per lo più Tal) 1 2 1 3 3 4 2. Mutazione della regione “promoter” 3. Anomalie dello splicing 4. Mutazione del segnale di poliadenilazione 5 6 5. Interruzione prematura (Mutazione non senso e frameshift) 6. Emoglobine instabili
ETEROGENEITA’ DELLE TALASSEMIE • DIFFERENTI DIFETTI MOLECOLARI • DIFFERENTI ALLELI • DIFFERENTI GENOTIPI • DIFFERENTI FENOTIPI
TRASMISSIONE DELLA BETA TALASSEMIA 25% Normale 50% Malato 50% 25% Portatore asintomatico
STATI CLINICO-EMATOLOGICI DELLA TALASSEMIA 1. PORTATORE “ASINTOMATICO” - TALASSEMIA MINOR o MINIMA (MICROCITEMIA) 2. TALASSEMIA INTERMEDIA 3. TALASSEMIA MAIOR (MORBO DI COOLEY)
TALASSEMIA MINOR eterozigosi • “Anemia” più frequente in Italia, con tipica diffusione in alcune zone (Italia meridionale, isole e zone delta padano) • Ridotta sintesi di catene • Diagnosi per lo più casuale, raramente moderati sintomi anemia dipendenti • Importanza della diagnosi a scopo eugenetico
TALASSEMIA MINOR-MINIMA Eterozigosi EMOCROMO NORMALE TALASSEMIA MINOR-MINIMA 15. 0 10 -15 5. 000 6. 000 HCT (%) 45 35 MCV ( 3) 90 60 Hb A 2 < 3% Hb A 2 3% Hb g/d. L Eritrociti x 106/ L Elettroforesi Hb
PRINCIPALI GENOTIPI E FENOTIPI EMOGLOBINICI NELLA TALASSEMIA MAIOR (MORBO DI COOLEY) GENOTIPO HB FENOTIPO HB Omozigosi tal 0 Prevalenza Hb. F Hb. A 2 aumentata Hb. A assente Omozigosi tal + Prevalenza Hb. F Hb. A 2 aumentata Hb. A presente Omozigosi -tal 0 Solo Hb. F Omozigosi Hb Lepore Hb. F + Hb Lepore Doppia eterozigosi per i difetti su elencati Variabile (sempre prevalente Hb. F)
PATOGENESI DELL’ANEMIA NEL MORBO DI COOLEY • Anemia grave per ridotta o assente sintesi delle catene globiniche e quindi di Hb. A • Le catene in eccesso precipitano nei precursori eritroidi causando eritroblastolisi endomidollare e ridotta sopravvivenza degli eritrociti circolanti
CLINICA DEL MORBO DI COOLEY ANEMIA • Rilievo, in genere entro i primi sei mesi di vita, di grave anemia con: – Pallore-colorito itterico – Splenomegalia: per iperplasia della polpa rossa secondaria all’esaltata eritrocateresi – Epatomegalia: persistenza post fetale di mielopoiesi extramidollare e per alterazioni del circolo epato-splenico – Insufficienza cardiaca • Ritardato sviluppo somatico e sessuale
CLINICA DEL MORBO DI COOLEY Epato-splenomegalia ingravescenti
CLINICA DEL MORBO DI COOLEY ALTERAZIONI OSSEE • Secondarie all’iperplasia del tessuto emopoietico nelle ossa spugnose • Alterazioni caratteristiche: facies simil-asiatica • Alterazioni radiologiche caratteristiche (per es. cranio a spazzola, osteoporosi) • Deformità articolari e prematura fusione delle epifisi
CLINICA DEL MORBO DI COOLEY Deformazioni facciali
CLINICA DEL MORBO DI COOLEY SOVRACCARICO DI FERRO Per: • Aumentato assorbimento di ferro • Carico emotrasfusionale • I sintomi si sviluppano soprattutto con l’età • Organi bersaglio: fegato, cuore, ghiandole endocrine (pancreas, paratiroidi, gonadi, ipofisi, tiroide) • Quadri clinici più invalidanti: insufficienza cardiaca, diabete, cirrosi epatica
CLINICA DEL MORBO DI COOLEY Cardiomegalia in paziente con Morbo di Cooley e sovraccarico marziale Sezioni post mortem di miocardio con depositi di ferro
LABORATORIO NEL MORBO DI COOLEY • Severa anemia (4 -6 g/d. L), ipocromica, microcitica, con marcata anisopoichilocitosi (schistociti, dacriociti e forme bizzarre) ed emazie a bersaglio • Presenza di eritroblasti ortocromatici nel sangue periferico • Leucociti e piastrine normali o ridotti per sequestro splenico • Mielobiopsia: iperplasia eritroblastica con eritropoiesi inefficace, aumento ferro emosiderinico • Prevalenza di Hb. F (pone la diagnosi definitiva) • Iperbilirubinemia indiretta (4 -8 mg/100 ml) • LDH aumentato • Sideremia e ferritinemia aumentate
TERAPIA DEL MORBO DI COOLEY • Terapia trasfusionale (target: Hb > 10. 511 g/d. L) • Terapia chelante del ferro: desferrioxamina e chelanti orali • Splenectomia • Trapianto di cellule staminali emopoietiche allogeniche • Terapia genica ?
TRAGUARDO DELLA TERAPIA TRASFUSIONALE • Attività normale • Accrescimento normale • Riduzione iperplasia midollare prevenzione delle alterazioni scheletriche • Riduzione dell’ipervolemia minor sovraccarico cardiaco • Riduzione della splenomegalia e del conseguente ipersplenismo
PREVENZIONE DEL MORBO DI COOLEY • Diagnosi delle condizioni di eterozigosi • Educazione • Diagnosi prenatale
TALASSEMIA INTERMEDIA • Omozigosi per difetti minori • Doppie eterozigosi • Minor difetto sintesi Hb • Meno eritropoiesi inefficace • Minor necessità di terapia trasfusionale • Meno complicazioni
Base genetica di thalassemia: chr. 11 • solo 2 geni beta • Diminuiti livelli di beta provocano anemia ipocromica e microcitica. Le catene alfa in eccesso precipitano • beta globina non è necessaria durante vita fetale: talassemia beta compare solo alcuni mesi dopo la nascita
thalassemia: il quadro d’insieme clinical severity lieve WT thal. minor gravissima + mut 0 Null + thal. + + thal. major Hb. A Scarsamente presente + 0 No Hb. A 0 0 0 thal.
I corpi di Heinz in un globulo rosso a forma di goccia di lacrima (teardrop)
Thalassemia: il quadro clinico thalassemia (Cooley's anemia): range ampio di gravità da quello di anemia microcitica lieve ( thal. trait or thal. minor) con lieve iperplasia midollare a grave anemia emolitica con ritardo dello sviluppo, ittero, epatosplenomegalia, e iperplasia midollare grave associata a deformità ossee Terapia Trasfusionale può recare sovraccarico di ferro thalassemia Non trattata Guancia prominente, protrusione della mascella per espansione della cavità midollare nelle ossa del cranio a causa della eritropoiesi inefficiente
Trattamento Trapianto di midollo osseo è l’unica terapia esistente. Altri trattamenti includono trasfusioni di sangue, terapia delle infezioni, e splenectomia
Distribuzione delle talassemie Il fattore etnico Thalassemias predominano tra popoli che discendono dal Mediterraneo, Africa, Medio oriente, India, China, e South Est Asia. Tratto thalassemia: da 0. 01% in UK, Islanda e Japan, a 49% tra inhabitants di talune isole del Southwest Pacifico Tratto thalassemia: da 1. 5% Africans e African. Americans a 30% in taluni comuni della Sardegna
Mutazioni che interessano i geni della emoglobina
Base genetica di thalassemia: Sopratutto mutazioni puntiformi transcription RNA splicing RNA cleavage frameshift * nonsense codon unstable globin cap site initiator codon small deletion Mutazioni nel promotore, alterano splicing e maturazione m. RNA, mutazioni nonsense. Causa molteplicità mutazioni, omozigoti per la stessa mutazione sono rari
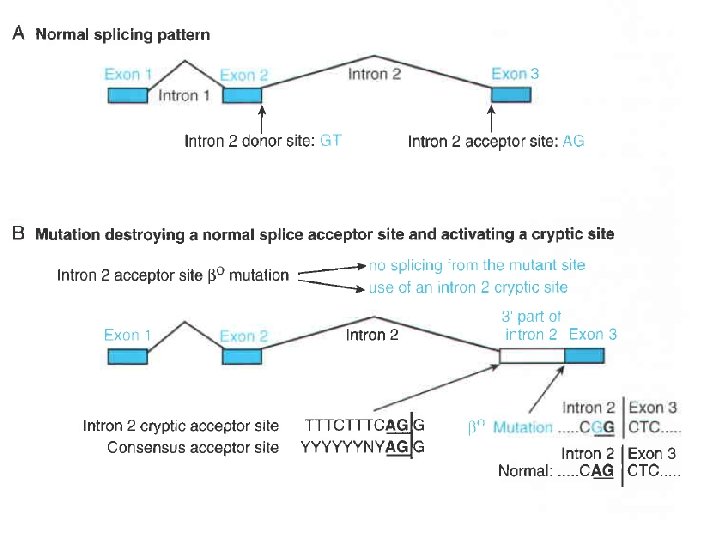
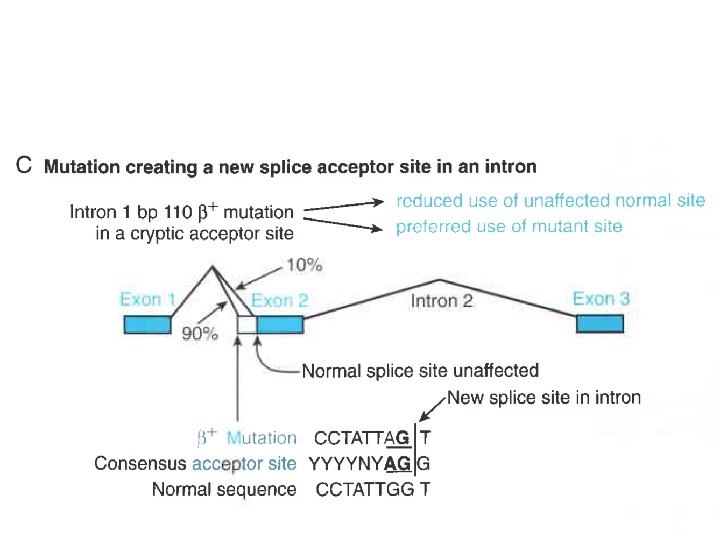
Mutazioni nelle regioni introniche una mutazione puntiforme che altera la sequenza GT alla estremita’ 5’ del grande introne (II) del gene codificante la beta-globina
Mutazioni nelle regioni introniche
Hb. E
HPFH
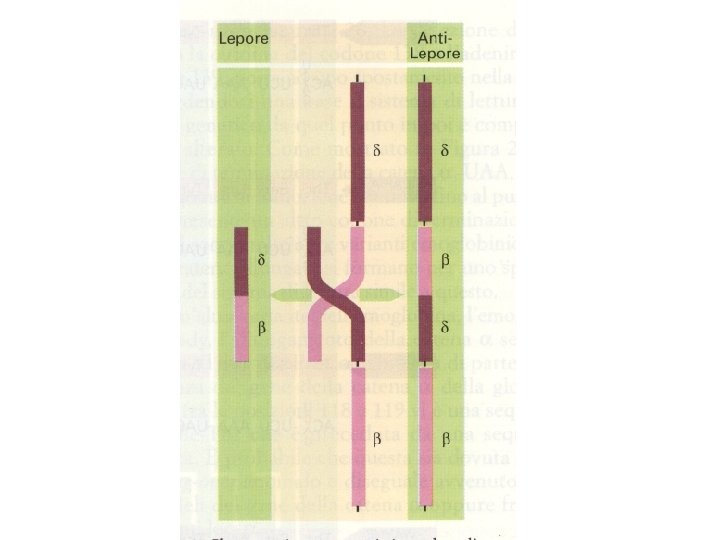
Fusioni e riarrangiamenti genici Hb Lepore Hb Kenia
Sono stati identificati vari tipi di Hb anti-Lepore: Hb Mjyada (conversione tra 12 e 22) Hb P-Nilotic (conversione tra 22 e 50)