Immune Deficiency Syndromes Immune deficiencies are divided into
































 Hyper – Ig. M Syndrome • X-Linked hyper-Ig.")
 X-Linked Lymphoproliferative Disease \"Duncan Syndrome\" \"XLP\" • Inability of T cells to regulate")

 Chediak-Higashi Syndrome It is an autosomal recessive disease characterized by abnormal giant granules")
 Chronic Granulomatous Disease \"CGD\" • The final step in killing of ingested organisms")
 Interferon- Receptor Deficiency • A mutation in the IFN RI gene results in")
 Deficiencies of Early Complement")


 Defective Control of Complement Components (Hereditary Angioedema) • Patients lack C 1 esterase")
 Glycosyl Phosphatidy Inositol protein Deficiencies • A family of proteins with glycosyl phosphatidyl")









: encodes the viral enzymes, reverse transcriptase, integrase, and protease. •")





 – Transplacental transmission –")


 Malignancies • Kaposi's sarcoma • Burkitt-like lymphoma • Hodgkin's disease • CNS lymphoma")
































- Slides: 103
Immune Deficiency Syndromes ØImmune deficiencies are divided into two major categories: o Primary immune deficiency, which may hereditary or acquired, in which the deficiency is the cause of disease. o Secondary immune deficiency, in which the immune deficiency is a result of other diseases or conditions.
Ø Categories of primary immune deficiencies: 1. T cell "cell-mediated immunity". 2. B cell "antibody-mediated immunity". 3. Both B and T cell immunity. 4. Nonspecific immunity mediated by phagocytic cells and/or NK cells. 5. Complement activation. Ø Immune deficiency should always be suspected in a patient with recurrent infections. Table 17. 1 p 234 coico shows the types of infections. For example, recurrent bacterial otitis media and bacterial pneumonia are common in individuals with B cell and antibody deficiency.
Ø Increased susceptibility to fungal, protozoan and viral infections is seen with T cell and cell-mediated immune deficiency. Ø Systemic infections with low virulence bacteria, superficial skin infections, or infections with pyogenic organisms suggest deficiencies in phagocytic cells. Ø Recurrent infections with pyogenic organisms are associated with complement deficiencies. Ø Pneumocystic carinii, cytomegalovirus, toxoplasmosis, Mycobacterium avium, and candida are the most common organisms associated with deficiencies in cell-mediated immunity.
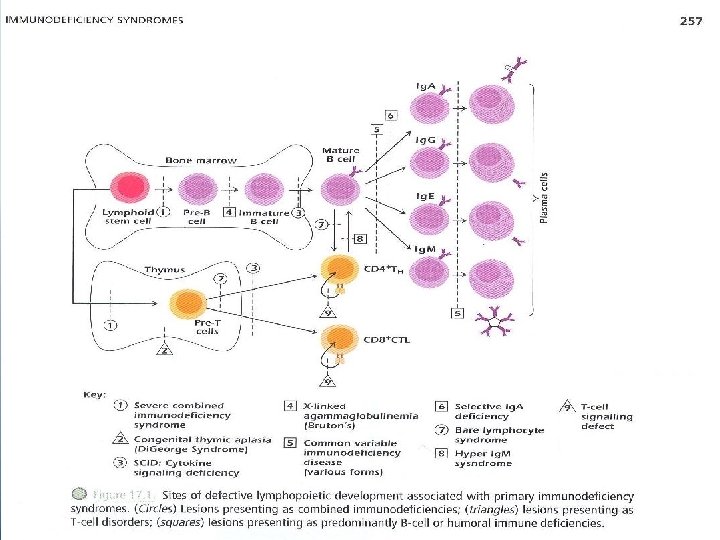
Primary Immunodeficiency Syndromes Ø With the exception of Ig. A deficiency, the frequency of primary immune deficiency syndromes is very low "1 in 10, 000"; 50% of all cases are antibody deficiencies 20% are combined deficiencies in antibody and cell-mediated immunity. 18% are phagocytic disorders 10% are disorders of cell=mediated immunity alone. Ø Figure 17. 1 shows that the earlier the genetic defect or block occurs in development, the more arms of the immune system are affected and the more severe the disease.
* Severe Combined Immunodeficiency Diseases Ø SCID comprises a heterogeneous group of diseases in which both cell-mediated immunity and antibody production are defective (Fig 17. 1). Ø SCID was originally called swiss-type agammaglobulinemia. Ø Individuals with SCID are susceptible to virtually every type of microbial infection "viral, bacterial, fungal, protozoal", most notably CMV, Pneumocystis carinii and candida. Vaccination with attenuated live virus could prove fatal in such infants. Ø Patients can be subclassified according to the lymphocyte subsets present in their blood "Table 17. 2”
o T– B+ "absent T cells, normal B cells", they may lack NK cells. o T– B– "both T and B cells are absent" o T+ B+ few patients. o T+ B– rare patients ØThe preferred treatment for all SCID patients is a T cell-depleted bone marrow transplant from an HLA-matched sibling donor.
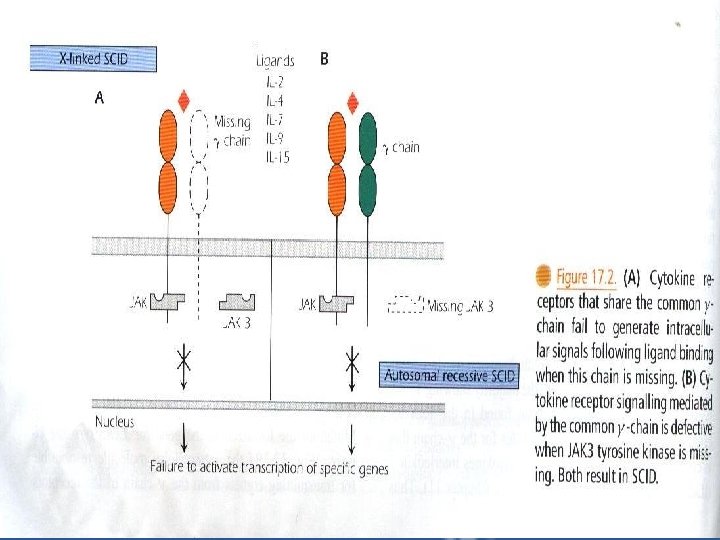
I. T– B+ Subgroup: a – X-Linked SCID which constitutes 40 -50% of cases with the majority showing T– B+ lymphopenia. – Mutations are found in the gene located on the X chromosome that codes for the -chain that is common to the receptors for IL-2, IL-4, IL-7, IL-9 and IL-15. thus the mutation impairs responses to a multitude of cytokines "Fig 17. 2 A”
b – Autosomal Recessive SCID – Patients have mutations localized in the gene for JAK 3 tyrosine kinase "Fig 17. 2 B", the intracellular molecule responsible for transmitting signals from Expression of JAK 3 is normally restricted to hematopoietic cells.
II. T– B– Subgroup a – Adenosine Deaminase Deficiency. Adenosine deaminase "ADA", an enzyme in the Purine Salvage Pathway. Individuals lacking this enzyme account for 20% of SCID patients and show an autosomal recessive pattern of inheritance. The deficiency results in build up of toxic wastes. ADA deficiency has greatest impact of the immune system resulting in failure of both T and B lymphocyte development.
b – Purine Nucleoside Phosphorylase Deficiency ne se enzyme, deficiency leads to a buildup of toxic products that are particularly damaging to the neurologic system and T cells. Eventually, all lymphoid tissues are depleted.
c – Recombinase Deficiency Recombination-activiting genes "RAG" 1 and 2 code for enzymes involved in the rearrangement of the Ig genes in pre-B cells and the T cell receptor genes in pre-T cells. Both enzymes are absolutely required for rearrangement. So mutation in either result in complete absence of T cells B cells, and Ig. Maturation stops at the pre. T and pre-B cell stages. NK cell function is intact.
III. T+ B– Subgroup Omenn Syndrome is a leaky SCID with reduced but partial RAG activity. Clinical presentations are similar to individuals with GVH disease rather than to those lacking RAG activity. Patients are severely immunodeficient and demonstrate a dystregulation of the immune system. They have massive skin and GIT infiltration by eosinophils and activated T cells producing TH 2 type cytokines, this results in hyper-Ig. E syndrome and malnutrition due to protein loss. Patients still need pretreatment with immunosuppressive therapy.
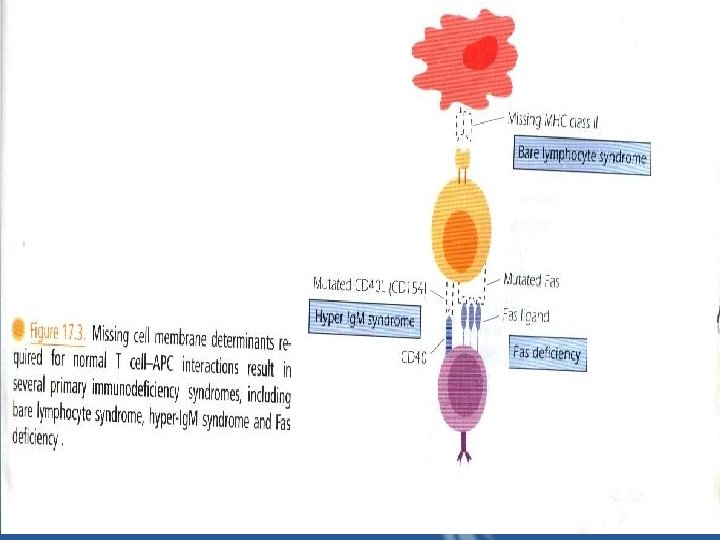
IV. T+ B+ Subgroup a – Bare Lymphocyte Syndrome "BLS" results from the failure to express HLA molecules. – only those individuals lacking expression of HLA class II molecules, with or without class I expression, consistently show immunodeficiencies. – Circulating T and B cells may be normal, however, in the absence of HLA class II molecules, protein antigens cannot be presented to CD 4+ T cells.
b – Zap-70 Mutation – Patients with a mutation in the T cell tyrosine kinase ZAP-70, which tranduces the signal transmitted through the T cell receptor, also present with a SCID like phenotype.
V. Other Multisystem Disorders – In addition to the SCID, several multisystem inherited disorders result in a SCID-like clinical picture. a – Wiskott-Aldrich Syndrome – X-Linked disease showing a classic triad of symptoms: § bleeding diathesis "tendency to bleed" due to thrombocytopenia. § recurrent bacterial infections. § allergic reactions.
§ Longer term, patients have increased risk of developing malignancies, particularly of the lymphoid system. § The genetic basis of the disease is a mutation in the X-Linked gene coding for the Wiskott. Aldrich Syndrome protein "WASP", which is expressed in all hematopoietic stem cells. § Both T and B cells are functionally abnormal, patients are unable to respond to polysaccharide antigens.
§ Treatment * antibiotics * antiviral agents * bone marrow transplantation - without treatment the average life span is approximately 3 years. b – Ataxia Telangiectasia "AT" – A multisystem genetic disorder includes: § lack of muscle coordination "ataxia" § abnormal vascular dilatation "telangiectasia" in the eyes and skin
§ increased susceptibility to infection § lymphopenia § immune defect involves both T and B cells ØThe gene basis of AT is a mutation in the gene coding for a protein known as ATM. Øchildren have risk of developing malignancy, particularly lymphoid neoplasms.
v. Immunodeficiency Disorders Associated with T Cells and Cell-Mediated Immunity Ø Patients with T cell-associated deficiency diseases are susceptible to viral, fungal, and protozoal infections. Ø T cells help B cells to produce antibodies to Tdependent antigens, patients with T cellassociated deficiencies also exhibit selective defects in antibody production. Consequently, T cell deficient patients may be difficult to distinguish clinically from SCID patients.
a- Congenital Thymic Aplasia "Di. George Syndrome" • Di. George Syndrome is a T cell deficiency in which the thymus develop abnormally. • The syndrome is caused by defective migration of fetal neural crest cells into the third and fourth pharyngeal pouches. • The heart and face develop abnormally, ant the thymus and parathyroids fail to form resulting in thymic aplasia and hypoparathyroidism.
• Thymic aplasia results in an absence of mature T cells and immunodeficiency. • Di. George Syndrome is not hereditary but occurs sporadically and is generally the result of a deletion in chromosome 22 q 11. • Patients suffer from recurrent or chronic infections with viruses, bacteria, fungi and protozoa. • Patients with Di. George syndrome lack T cells and fail to generate normal antibody responses, they should never be immunized with live attenuated vaccines.
b- T-cell Deficiencies with Normal Peripheral T cell Numbers • Functional defects in T cells. • Opportunistic infections and high incidence of autoimmune disease are observed. • Autosomal recessive patterns of inheritance. • The underlying cause is heterogenous, with deficient expression of ZAP-70 tyrosine kinase, CD 3 E, or CD 3 , these molecules are involved in T cell signaling through the antigen-specific receptor "TCR".
c- Autoimmune Lymphoproliferative Syndrome "ALPs" • It is an inherited disease characterized by massive proliferation of lymphoid tissue with early lymphoma development. • It may be considered as autoimmune disease with susceptibility to chronic viral infections. • Patients have an increased number of double negative "CD 4–CD 8–" T cells and may develop B cell lymphoma.
• Most ALPs patients have a mutation in the gene coding for the Fas protein "CD 95" "apoptosis signaling". • Most ALPs patients have one normal and one mutated Fas molecule. • This suggests that the mutated Fas molecule interferes with the function of the normal molecule. • Some ALPs patients have defects in other components of the apoptosis pathway, such as Fas ligand or caspase 10.
d- Chronic Mucocutaneous Candidiasis • Characterized by candida infections of skin and mucous membranes. • Patients usually have normal T cellmediated immunity to microorganisms other than candida and normal B cellmediated immunity to all microorganisms including candida. They have selective defect in the functioning of T cells.
*B cell or Immunoglobulin – Associated Immunodeficiency Disorders a-X-Linked infantile Agammaglobulinemia "Bruton's agamma-globulinemia" • Infants present with serious and repeated bacterial infections as a result of severe depression or virtual absence of all Ig classes. • The major defect lies in the inability of pre-B cells to develop into mature B cells.
• The BTK gene, which is mutated in XLA, normally codes for a tyrosine kinase enzyme residing in the cytosol. • XLA is inherited immunodeficiency disease in which a mutation in a cytoplasmic tyrosine kinase is responsible. • Patients suffer repeated infections. • Treatment consists of periodic injections of intravenous gamma globulin "IVGG" containing large amounts of Ig. G.
b- Transient Hypogammaglobulineamia • At 5 -6 months of age, passively transferred maternal Ig. G disappears and production by the infant begins to rise. • Premature infants may have transient Ig. G deficiency if they are not yet able to synthesize Ig. Sometimes helper T cells are deficient. • This disease may persist for few months to as long as 2 years. • It is not X-Linked and can be distinguished from XLinked disease by the presence of normal numbers of B cells. • Immunization should not be given during this period.
c- Common Variable Immunodeficiency Disease "CVID" • CVID is characterized by decreased serum Ig. G and Ig. A with normal or low Ig. M and normal or low peripheral B cell numbers. • B cells fail to mature into plasma cells. • Affected individuals suffer from recurrent respiratory and gastrointestinal infections. • Treatment with IVGG.
*Selective Immunoglubolin Deficiencies - Ig. A deficiency • Patients may suffer from recurrent viral or bacterial infections, celiac disease "defective absorption in the bowel", or may be entirely asymptomatic. • Treatment with broad spectrum antibiotics. - Ig. M Deficiency • It is a rare disorder. • Patients suffer from recurrent and severe infections with polysaccharide encapsulated organisms, such as pneumococci and Haemophilus influenzae.
* Disorders of T-B Interactions a) Hyper – Ig. M Syndrome • X-Linked hyper-Ig. M Syndrome "XHIM" present with recurrent respiratory infections at 1 -2 years of age. • A mutation in the CD 40 L gene on the X chromosome results in the absence of CD 40 Ligand "CD 154" on TH cells "Fig 17. 3 coico p 241". CD 40 L binds to CD 40 expressed on B cells this interaction rescues the B cell from apoptosis.
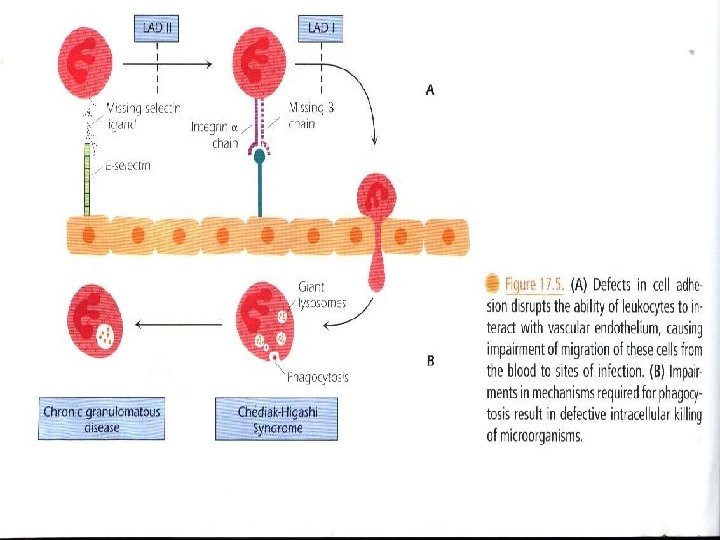
b) X-Linked Lymphoproliferative Disease "Duncan Syndrome" "XLP" • Inability of T cells to regulate B cell growth is considered to be a major part of the underlying defect in this rare disease. • The lymphoma incidence is high in XLP. * Phagocytic Dysfunction a) Leukocyte Adhesion Deficiency "LAD" LAD is a group of disorders in which leukocyte interaction with vascular endothelium is disrupted "Fig 17. 5 “
• LAD I is an autosomal recessive disease mapping to chromosome 21, patients have a defect in the subunit of integrin molecules, preventing their expression. As a result, adhesion and migration of all white blood cells are impaired. • LAD I individuals suffer from recurrent soft tissue bacterial infections and have increased WBC counts but without pus formation or effective wound healing. Lymphocyte function is also affected due to the lack of LFA-1 expression. • LAD II – Individuals have a defect in selectin ligand. The underlying defectin LAD II is in fucose metabolism, which results in the absence of fucosylated ligands for selectins to bind to.
b) Chediak-Higashi Syndrome It is an autosomal recessive disease characterized by abnormal giant granules and organelles in the cell "Fig 17. 5. B". Neutrophils show diminished intracellular killing of organisms, the result of both defective degranulation and impaired function of lysosomes with phagosomes. • Massive infiltration of lymphocytes and macrophages in the liver, spleen and lymph nodes. • Pyogenic organisms "strept, staph" cause recurrent fatal infection.
c) Chronic Granulomatous Disease "CGD" • The final step in killing of ingested organisms is defective "Fig 17. 5 B" and the continued intracellular survival of the organisms results in granuloma formation. • Mutations in any enzyme that catalyzes the respiratory burst can result in CGD. • The intracellular killing of organisms is decreased. • Symptoms appear during the first 2 years of life. Patients suffer infection with low virulence organisms.
d) Interferon- Receptor Deficiency • A mutation in the IFN RI gene results in an inability of monocytes to respond to IFN with secretion of tumor necrosis factor- "TNF- ". • Patients are susceptible to weakly pathogenic mycobacteria. * Natural-Killer Cell Deficiency • NK cell deficiency impairs allograft rejection and is linked to higher susceptibility to viral diseases and increased metastases from tumors. • NK deficiency are seen in SCID, some T and phagocytic cell disorders, and in X-Linked lympoproliferative syndrome.
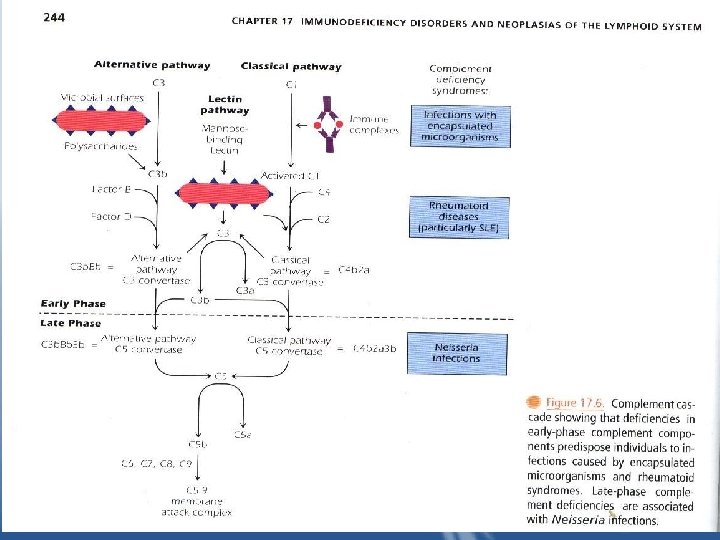
* Diseases Caused by Abnormalities in the Complement System a) Deficiencies of Early Complement Components • The early components of complement are important in generating the opsonin C 3 b "Fig 17. 6”
• Patients with deficiencies of C 1, 4, or 2 from the classical pathway or with C 3 deficiency itself have increased infections with encapsulated organisms "S. pneumoniae, S. pyogenes, H. influenzae" and increased rheumatic diseases due to improper clearing of immune complexes as a consequence of low C 3 b generation. • SLE is the most common presenting symptoms of some complement deficiencies. • Deficiencies of the mannose binding lectin, which binds to the surface of microbes without antibody and activates the classical pathway, also result in risk of bacterial infections and SLE-like symptoms.
• Deficiency of C 3 itself is associated with most severe symptoms particularly infections complications. b) Deficiencies of Late Complement Components • Deficiencies of c 5, C 6, C 7, C 8, C 9 interfere with the generation of the membrane attack complex (MAC). The MAC is directly lytic and the primary defense against gram-negative bacteria, particularly N. menigitidis.
c) Defective Control of Complement Components (Hereditary Angioedema) • Patients lack C 1 esterase inhibitor. The action of C 1 on C 4 and C 2 and the kallikrein system is uncontrolled, generating large amounts of vasoactive peptides patients suffering from localized edema which is life threatening when it occurs in the larynx, obstructing the airway passage. • Treatment includes avoidance of trauma, and infusion of C 1 esterase inhibitor.
d) Glycosyl Phosphatidy Inositol protein Deficiencies • A family of proteins with glycosyl phosphatidyl inositol "GPI" anchors is expressed on the membranes of RBCs, lymphocytes, granulocytes, endothelial cells and epithelial cells. These proteins include decay-accelerating factor "DAF, or CD 55" and CD 59 protect the cells against spontaneous lysis by complement. In the absence of these cell surface inhibitors, granulocytes, platelets and, particularly RBCs are susceptible to spontaneous lysis by complement.
• An acquired form of the disease, called paroxysmal nocturnal hemoglobinuria "PNH" is common. In PNH, patients have a deficiency in an enzyme required for the production of all the GPI anchored proteins. This is due to an acquired somatic mutation in an early myeloid stem cells. • During the chronic course of PNH, intravascular hemolysis occurs in the kidney's at night where the acidic environment activates the alternative complement pathway.
Secondary Immunodeficiency Diseases • They are the consequence of other diseases. • The most common cause worldwide is malnutrition. • Immunodeficiency is more often iatrogenic-caused by medical treatment – particularly from using chemotherapeutic agents in cancer therapy or deliberate immunosuppression in cases of organ transplantation or autoimmune diseases. • Secondary immunodeficiencies are also seen in untreated autoimmunity and with overwhelming infections by bacteria. • Malignancies of the immune system also frequently suppress the nonmalignant components, resulting in increased susceptibility of these patients to infection.
Acquired Immunodeficiency Syndrome "AIDs" •
• The etiologic agent is the Human Immunodificiency virus "HIV". • The causative agents of acquired immune deficiency syndromes (AIDS). • They belong to the lentivirus subfamily of the Retroviridae family. • The genome is diploid, comprising two singlestranded RNA molecules. • HIV exhibits a characteristic cone-shaped core. Figures 1 a & b represent the main components of the virus. • HIV is enveloped "lipid bilayer derived from the host cell membrane".
• The viral envelope is studded by virally encoded envelope glycoproteins: – gp 120 (surface unit) – gp 41 (transmembrane unit) • other proteins in the virus particle – P 17 (matrix protein), lines the inner surface of the virus particle. – P 24 (major capsid protein), surrounds two copies of RNA. – P 9 (nucleic acid binding protein). • Protease – Integrase – Reverse transcriptase In the capsid structure
• It is estimated that a single HIV-1 virion contains about 1200 molecules of P 24, 80 molecules of reverse transcriptase, 280 molecules of gp 120. • HIV virions also contain a number of cellular proteins including beta-2 microglobulin, major histocompatibility complex (MHC) antigens, actin and ubiquitin. • These proteins, together with gp 120 surface glycoprotein, form "adhesion patches" that facilitate virus/cell fusion and core penetration. • More important, these proteins have also been implicated in viral pathogenesis, e. g. , virion associated class II molecules may trigger T cell receptor signaling on target cells causing their death.
• Cellular Targets of HIV Infection • Infection – CD 4+ T-lymphocytes – CD 4+ Monocytes and macrophages including microglia – CD 4+ Dendritic cells, including langerhans cells • Attachment – Folicular dendritic cells in lymph nodes – M cells on Peyer's patches – Galactosylcerebroside positive cells in brain and gut
• CD 4=gp 120 Interactions • The major cell surface receptor for HIV is the CD 4 differentiation antigen. • Several chemokine receptors act as coreceptors for HIV. • Macrophage tropic HIV uses the chemokine receptor CCR 5, and requires only a low level of CD 4 expression on the host cells. • Lymphotropic HIV uses the chemokine receptor CXCR 4 expressed on T cells and requires high density of CD 4 on the cell surface. • Genomic Organization of HIV
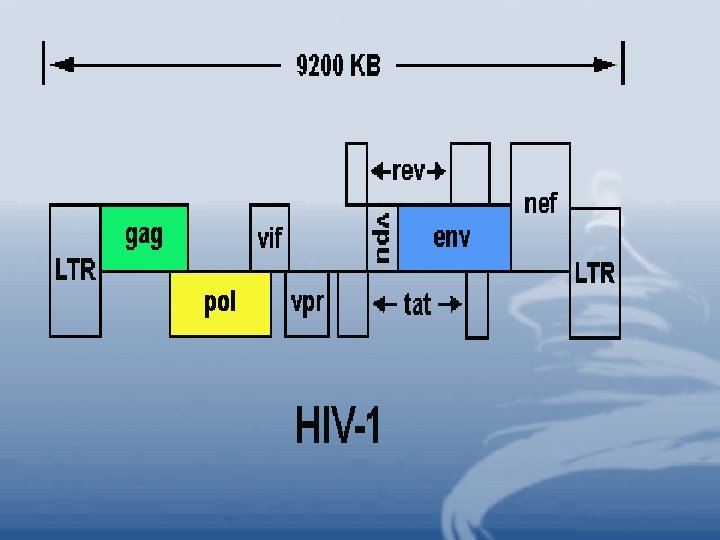
• The 9. 6 kb genome of HIV consists of both structural and regulatory sequences (Figure 3). Figure 4 illustrates the essential points in the HIV life cycle. • HIV Genetic Composition • Structural Genes • env (envelope): encodes the viral envelope proteins; gp 120 and pg 41. • gag (group associated antigen) encodes the viral proteins: p 24; capsid "CA" (core shel protein and p 17 "MA" (membrance associated matrix protein.
• pal (polymerase): encodes the viral enzymes, reverse transcriptase, integrase, and protease. • gag and pol genes cooperate to form fusion proteins which cause fusion of cells enabling HIV to pass from one cell to another thus evading the components of the immune system. • Long terminal repeats (LTR) at both 3` and 5` ends contain enhancer and promoter elements necessary for transcription. • HIV is initially transcribed into a full-length m. RNA, which is translated into the structural Gag and Pol proteins. • Single and spliced m. RNA is necessary for the synthesis of the envelope proteins.
• Regulatory Genes • tat: ”transactivator of transcription” regulates HIV replication and accelerates viral protein production by a thousandfold. • rev: ” regulator of virion” permits unspliced m. RNA to leave the nucleus, it inhibits transcription of the regulatory genes while enhancing expression of the viral structural genes. • vif: “viral infectivity factor” increases viral infectivity and may be responsible for the efficient cell-to-cell transmission of HIV.
• nef: “negative regulatory factor” enhances HIV replication and makes the virus more pathogenic. • vpr: ”viral protein R” plays a role in the regulation of viral and cellular gene expression and may be important in viral assembly and infection of macrophages, it may also induce apoptosis. • vpu: “viral protein U” influences release of HIV from the cell surface and contributes to CD 4 degradation within the endoplasmic reticulum.
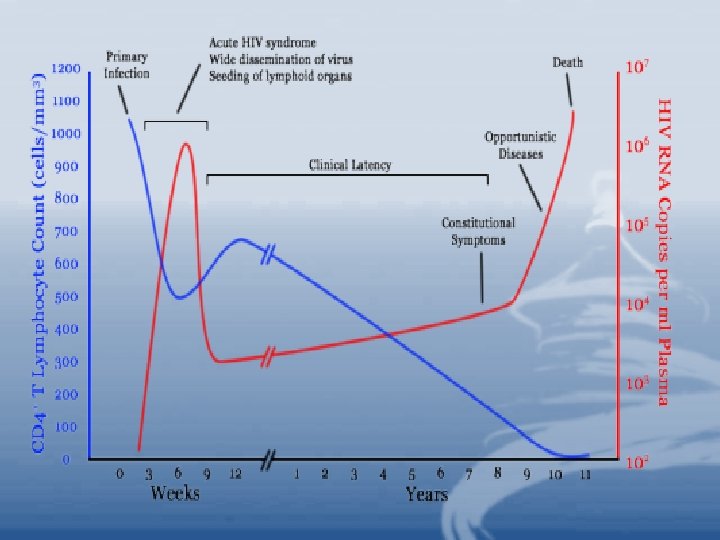
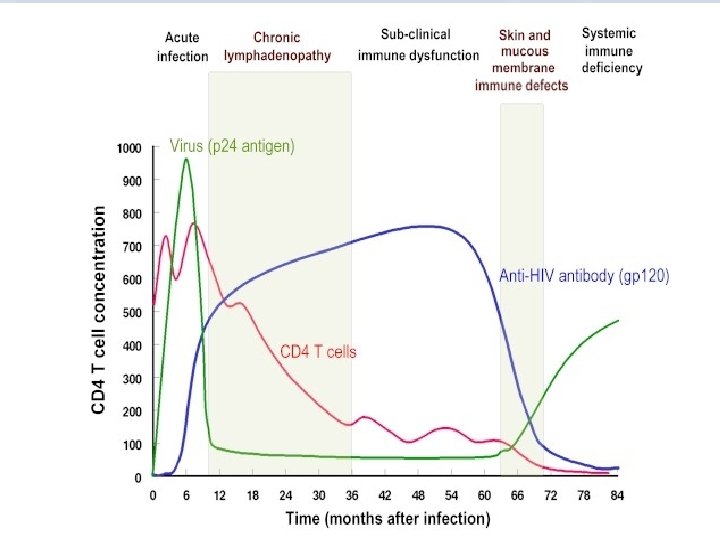
• HIV Infections and Pahtogenesis • During the early phase of HIV infection before seroconversion, the virus propagates mainly in peripheral blood mononuclear cells (PBMC). • Primary infection may cause a temporary depletion of CD 4+ PBMCs.
• HIV infection usually elicits strong cellmediated immune responses (CD 8+ cytotoxic T cells) and humoral responses help to clear virus load but fail to eradicate HIV infection. • During asymptomatic period, the virus is not completely latent but remains active in lymphoid tissue as proved by PCR technique. • Monocytes are resistant to destruction by HIV, they may act as a reservoir for the virus, as well as, a vehicle which transmits HIV to distant parts of the body.
• Epidemioloyg of HIV Infection • It is estimated that more than 40, 000 persons were infected with HIV form the beginning of the pandemic until the year 2006. In sub-saharan Africa, 25. 3 million are living with AIDs. . , in 2006, 4. 3 millions were infected, 3 millions died during the year 2006. more than 25 millions died since 1981.
• Modes of Transmission – Sexual contact (homosexual heterosexual) – Transplacental transmission – Drug injection – Percutaneous occupational exposure – Receipt of infected blood – Receipt of contaminated organs or tissues – Breast feeding • Common Opportunistic Infections Malignancies Associated with Aids A) Oppotunistic infections and
• Bacteria – Mycobacterium species – Streptococcus pneumonia – Haemophilus influenza – Salmonella species • Viruses – Herpes simplex – Herpes zoster – Epstein-Barr virus – Adenovirus – Cytomegalovirus
• Fungi – Candida albicans – Histoplasma capsulatum – Cryptococcus neoformans – Aspergillus species – Coccidioides immitis • Protozoa – Toxoplasma gondii – Pneumocystis carnii – Isospora belli – Cryptosporidium species
b) Malignancies • Kaposi's sarcoma • Burkitt-like lymphoma • Hodgkin's disease • CNS lymphoma • Immunoblastic sarcoma • Peripheral organ lymphoma • Lymphatic pre-leukemia
• Effect of HIV on the Immune Response – Depletion of CD 4 lymphocytes – Decreased delayed-type hypersensitivity – Increased susceptibility of opportunistic infections – Decreased production of IL-2 – Polyclonal hypergammaglobulinemia "Polyclonal B cell activation" – Production of autoantibodies and immune complexes – Unresponsiveness to new antigens – Decreased Natural Killer (NK) cell activity – Monocytes and macrophages chemotaxis inhibition
– Increased release of IL-1 – Increased circulating -IFN – Increased serum and urine neoprotein – Increased 2 microglobulin – Loss of HIV-specific cytotoxic T cells – Increased unresponsive CD 8+ T cells • Prevention and Control
• * Health education – Mass media campaigns – Adopt low risk sexual behavior – Provision of condoms – Tracing sexual partners – Targeting vulnerable groups – Prevention of the exchange of needles
* Early diagnosis * Blood supply testing * Early therapy * Counseling services * Special attention to demographic features of the epidemic; – Housing – Prisons – Rural healthcare – Immigration and travel
• Prevention HIV Infection in Health Care Workers – Wear coats and gloves when processing patient specimens. – Use masks and eyewear if splashing or aerosolization is anticipated. – Use precautions when handling needles and lancets. – Decontaminate work surfaces with a chemical germicide, e. g. , chlorox. – Wash hands and remove protective clothing before leaving work place. – Dispose of contaminated materials in autoclavable bags. – Avoid contaminating the outside of containers upon specimen collection-the lid should be tight.
Targets for Antiviral Therapy Target Therapy HIV binding and cell entry Soluble recombinant CD 4, dextran sulfate, castanospermine, heparin, soluble gp 120 Reverse transcription Zidovudin, Didanosine, Zalcitabine, Lamivudine, Stavudine, Nevirapine
Target Therapy HIV binding and cell entry Soluble recombinant CD 4, dextran sulfate, castanospermine, heparin, soluble gp 120 Reverse transcription Zidovudin, Didanosine, Zalcitabine, Lamivudine, Stavudine, Nevirapine Target Therapy HIV binding and cell entry Soluble recombinant CD 4, dextran sulfate, castanospermine, heparin, soluble gp 120 Reverse transcription Zidovudin, Didanosine, Zalcitabine, Lamivudine, Stavudine, Nevirapine
• Resistant strains of HIV-1 to many drugs have emerged. • A combination of antiviral drugs is recommended to overcome resistant strains. • Combined therapy may include "Highly Active Antiretroviral Therapy; HAART", for example: – one protease inhibitor plus two reverse transcriptase inhibitors. – Three reverse transcriptase inhibitors.
• Gene Therapy – Inhibition of viral translation process using anti-sense oligonucleotides or ribozyme. – Production of mutated proteins which compete with original viral proteins. – Genetic manipulation of target cells which enable them to be resistant for viral infection. – - Two DNA plasmids mixed together containing 5 HIV genes/ or Vaccinia virus carrying the same genes.
• Problems of developing effective vaccine – Antigenic diversity due to rapid mutation rate and recombination – Cell-cell fusion allows HIV to escape anti-HIV antibodies – Latent phase "integration into cellular DNA" – Cell-mediated immunity is deficient or delayed – Lack of animal model, e. g. , Chimpanzee developed antibodies but not AIDs – Anti HIV antibodies may react with CD 4 in a similar way to HIV-CD 4 interactions
– Auto-antibodies and immune complexes have harmful effect on many cells. – -Superinfection with other HIV strains. – -Binding of Abs to gp 120 is difficult. – Limited knowledge of mucosal immunity. – -Live attenuated vaccine may revert to virulence. – New Approaches: – Antibodies against HIV envelope proteins may neutralize infection – Addition of CD 4 molecules to cells may block the spread of HIV binds CD 4 preventing HIV from interaction with cellular-bound CD 4
• Assays for the Diagnosis of HIV infection – Serological screening – Antigen detection – Cell culture – Gene amplification Reaction, PCR" – CD 4 count "polymerase • Serological Screening – Detects circulating anti-HIV antibodies Chain
– ELISA … must be confirmed by: • Immunofluorescence • Radioimmunoprecipitation, or • Western blot [detects p 24, gp 41, gp 120] • PCR
2 - Neoplasms of the Lymphoid System • Lymphoid leukemias and lymphomas were originally categorized by cell morphology and clinical outcome. • A leukemia implies that the malignant cells are predominantly in the circulation and/or bone marrow. • A lymphoma presents as solid masses in the lymph nodes, spleen, thymus or extranodal organs. • Sometimes the same malignant cell type can show either presentation "leukemia/lymphoma".
• In 1996, the world Health Organization "WHO" recommended a classification system based on cell of origin – BVS T/NK- and stage of differentiation – immature "precursor" vs mature "peripheral" "Table 17. 4” • Malignant cells have the same surface markers but they may not continue to mature, accumulate in large numbers, and all originate from a single clone "monoclonal". They occupy the same site as their normal counter parts – bone marrow for immature B cells, thymus for immature T cells.
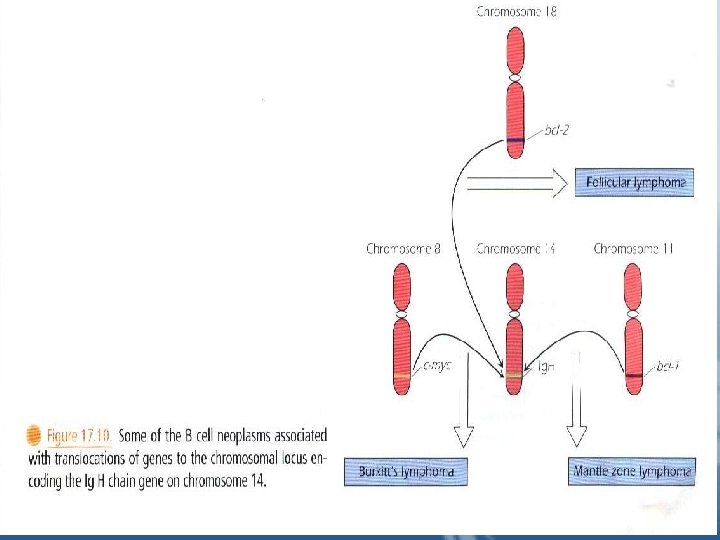
B Cell Neoplasms 1. Precursor B cell Lymphoblastic Leukemia /lymphoma – B cell acute lymphoblastic leukemias "B-ALLs" Correspond to the pro-B, pre-B, or immature B cell stages of development, as demonstrated by the expression of surface CD markers and the stage of Ig gene rearrangement in the individual patient's leukemia "Fig 17. 10”
• The malignant cells may express the blast or stem cell marker CD 34, and will express CD 10 and CD 19, early B cell markers. • ALLs express terminal deoxynucleotidyl transferase "Td. T" in their nucleus. The expression of this enzyme, normally required to rearrange the Ig genes "and T cell receptor genes" reflects the fact that B-All cells are in the process of gene rearrangement. • Chemotherapy has been successful in treating children with these leukemias.
2. Burkitt's Lymphoma/Leukemia It is characterized by a translocation that places the cmyc oncogene next to either the Ig. H chain gene or one of the two L chanin genes – t"I; 14", t"I; 22", or t"2, I" – “Fig 17. 10”. The c-myc protein activates genes for cell proliferation. • Translocation to the Ig genes leads to increased expression of c-myc and increased cell proliferation. • In equatorial Africa, this lymphoma is endemic, and is associated with EBV infection of the B cells.
• Burkitt's lymphoma is one of the malignancies seen in immunosuppressed patients in which EBV genome is also sometimes found in the lymphoma. 3. Follicular Lymphoma • It represents transformation of the B cells normally found in lymph node follicles. • In follicular lymphoma, the bcl-2 gene, which produces a protein that interferes with apoptosis, is translocated to the Ig. H chain gene "Fig 17. 10". This results in continuous expression of bcl-2 protein preventing death of the cells.
4. Mantle Cell Lymphoma • The normal germinal center of the lymph node is surrounded by a collar of small quiescent B cells that have not responded to antigen. A neoplasm of these mantle zone cells has the same B cell phenotype as their normal counterpart, CD 19+, CD 20+, CD 5+, s. Ig. M. • Many mantle cell lymphomas have a translocation of the bcl-1 gene to the Ig. H chain gene-t"11; 14" – resulting in overexpression of cyclin D 1 protein which promotes cell cycle progression form G 1 to S phase, leading to cell division. • Mantle cell lymphoma has a higher proliferative rate and a more aggressive course than follicular lymphomas.
5. Marginal zone lymphomas • They are common in the mucosal-associated lymphoid tissue "MALT", and, may be associated with chronic antigenic stimulation or autoimmune disease of that organ. • For example, chronic Helicobacter pylori infection of the stomach may lead to the development of gastric lymphoma and thus may be preventable by antibiotic treatment for the organism.
• Patients with autoimmune thyroiditis "Hashimoto thyroiditis" have a high incidence of this B cell lymphoma developing in the affected organ. • Chronic antigenic stimulation provides fertile ground for the development of a B cell lymphoma. B cells continue to undergo somatic mutation of their Ig genes. • Defect in the regulation of the B cells due to lack of T cell downregulation leads to both autoimmune disease and eventually lymphoma.
6. Chronic Lymphocytic Leukemia "CLL" / Small Lymphocytic Lymphoma "SLL" • A malignant transformation of B-1 cells. • B-CLL is the most common leukemia in North America and Western Europe, seen mostly in older individuals. • Patients are susceptible to infection. • Autoimmune antibodies are common particularly against RBCs, resulting in autoimmune hemolytic anemia. 7. Diffuse Large B cell Lymphoma • It may be a progression of other lymphomas e. g. follicular lymphoma, or may be a consequence of a poorly controlled EBV infection in immunosuppressed individuals.
8. Plasma Cell Neoplams • Neoplastic growths of plasma cells may occur at a single site, resulting in a plasmacytoma, or may be at multiple sites, predominantly throughout the bone, and are called multiple myeloma or plasma cell myeloma. IL-6 functions as an autocrine growth factor for myeloma cells. • The neoplastic plasma cells synthesize monoclonal proteins e. g. Bence-Jones Proteins "Light Ig chain", passes into urine. • Light chain deposits called amyloid can cause organ failure, especially in the kidneys.
9. Lymphoplasmacytic Lymphoma "Walclenstrom" Macroglobulinemia • It is a neoplasm of a single clone of B cells in which their microscopic appearance is a mixture of lymphocytes, plasma cells, and something in between-lymphoplasmacytoid. • There is overproduction of Ig. M. • The Ig. M has an abnormal structure, causing it to precipitate in the cold "cryoglobulin" and cause circulatory problems in their extremities "fingers and toes".
T – cell Neoplasma 1. Precursor T cell Acute Lymphoblastic Leukemia / T-ALL" Lymphoma • T-ALL is a neoplasm of immature T cells and has characteristics identical to those of thymocytes frozen in their immature state. • T-ALLs express the pan-T cell markers, CD 2, CD 5, CD 7. • Some T-ALLs do not express CD 4 or CD 8. others are double positive. • TCR have not completed rearrangement, and still express Td. T. • T-ALL presents as a leukemia or as a thymic mass.
2. Peripheral T – cell Neoplasms • They are more aggressive course than B cell lymphomas. A. cutaneous T cell lymphoma • infiltration of the epidermis by malignant CD 4+ T cells, eventually, the cells spread to the lymph nodes and even into the blood. • The malignant T cells found in the circulation are called Sezary cells and the patient is said to have Sezary Syndrome.
B. Adult T cell Leukemia "ATLL" / Lymphoma • • It is an aggressive T cell neoplasm. ATLL is a neoplasm of mature CD 4+ T cells. IL-2 is an autocrine growth factor for these cells. ATLL is caused by the retrovirus human T cell lymphotropic virus 1 "HTLV-1", which was described and isolated before recognition of AIDS and HIV.
• Transmission of HTLV-1 is similar to HIV, the incubation period is 20 -40 years. It infects CD 4+ T cells and nervous system. • About 1% of infected patients develop ATLL. CD 4+ T cells harbor the virus in a quiescent state. It is not cytolytic.
Hodgkin Lymphoma • It is characterized by the presence of relatively small numbers of large binucleate malignant cells named Reed-Sternberg Cells in a reactive background of small T cells, eosinophils, plasma cells, macrophages and fibrosis. This reactive milieu is the result of abundant cytokine production, particularly, IL-5 by the tumor cells and/or the background cells.
• Patients present with fever, night sweat weight loss, depressed cell-mediated immunity with increased susceptibility to viral and parasitic infections. • Recent studies using molecular techniques on single malignant cells have shown rearrangement of Ig genes suggesting that they are of B cell origin. • However, this lymphoma behaves differently from large B cell lymphomas and therefore remain separately classified. • Lymphomas are grouped as Hodgkin versus non. Hodgkin lymphoma.