COAGULATION DEFICIENCIES Severe coagulation deficiencies or coagulopathies typically


















![• Cryoprecipitate (the precipitate remaining after fresh-frozen plasma [FFP] is thawed at 4°](https://slidetodoc.com/presentation_image/78605de20af6f93e8d11ac7dcdf192c6/image-20.jpg "• Cryoprecipitate (the precipitate remaining after fresh-frozen plasma [FFP] is thawed at 4°")
- Slides: 20
COAGULATION DEFICIENCIES Severe coagulation deficiencies, or coagulopathies, typically are characterized by the development of excessive bleeding or bruising that is unprovoked or, more commonly, is precipitated by trivial incidental or surgical trauma. Frequently, these hemorrhagic events result in life- and limb-threatening complications. Moderate and mild coagulopathies may remain clinically silent until they are detected serendipitously on routine laboratory screening assays for global coagulation (e. g. , prothrombin time [PT] or activated partial thromboplastin time [a. PTT]) or when these assays are ordered to evaluate the cause of abnormal bleeding or easy bruisability.
• Normal Hemostasis Henry M. Rinder Normal hemostasis involves the physiologic balance of procoagulant and anticoagulant factors that maintains liquid blood flow and the structural integrity of the vasculature. Vascular damage results in initiation of clotting with the goal of producing a localized platelet/fibrin plug to prevent blood loss; this is followed by processes that lead to clot containment, wound healing, clot dissolution, and tissue regeneration and remodeling.
• In healthy persons, all these reactions occur continuously and in a balanced fashion such that bleeding is contained, but blood vessels remain patent and deliver adequate organ blood flow. When one or several of these processes are disrupted because of inherited defects or acquired abnormalities, disordered hemostasis may result in either bleeding or thromboembolic complications
• Much of the morbidity of coagulopathies can be minimized or avoided altogether by advanced awareness and prophylactic replacement of the deficient clotting factor proteins. In contrast to the lifelong clinical manifestations of hereditary or congenital coagulopathies, acquired deficiencies usually appear acutely in previously asymptomatic individuals, may not be suspected immediately on examination, and may remit spontaneously or after eradication of an inciting disease state or the withdrawal of an offending medication. Acquired coagulation disorders often are associated with more severe bleeding than are those derived from congenital causes. Coagulopathies predominantly result from inadequate biosynthesis of coagulation factor proteins or from direct or indirect inhibition of activated clotting factor proteins by acquired antibodies or anticoagulant medications; however, qualitative defects, either congenital or acquired, also can result in bleeding.
• The adhesive interactions producing stable platelet attachment to subendothelial von Willebrand factor (v. WF). The initial attachment between glycoprotein Ib (GPIb) and its binding domain on v. WF is rapid but has a short half-life, and the result is a rolling movement from torque generated by flowing blood. The v. WF-GPIb interaction produces transmembrane signaling that activates the platelet and transforms GPIIb/IIIa into a conformation capable of binding to the arginine-glycine-aspartate (RGD) domain on v. WF. This secondary adhesion is nearly irreversible and anchors the platelet to the exposed subendothelium. EC = endothelial cell
• The coagulation cascade. The in vitro "extrinsic" and "intrinsic" pathways converge to a common procoagulant pathway that results in thrombin generation and fibrin clot formation. As noted in the text, there is clear evidence that in vivo clotting function does not occur through these artificially distinct pathways, but the current clotting assays are best understood with this schema. The ability of thrombin to amplify its own formation by positive feedback on factor activation is demonstrated by the dotted lines. HMWK = high-molecularweight kininogen; PK = prekallikrein; TF = tissue factor
• Hereditary Hemophilias • Definition • The hemophilias include hemophilia A, caused by a deficiency of clotting protein factor VIII (antihemophilic factor), and hemophilia B, caused by a deficiency of factor IX (also called antihemophilic factor B, plasma thromboplastin component, or Christmas factor, named after an individual with the disease). A deficiency of either of these two intrinsic coagulation pathway components results in inefficient and inadequate generation of thrombin.
• Epidemiology • The sex-linked recessive disorders of hemophilia A and B are estimated to occur, respectively, in approximately 1 of every 5000 and 1 of every 30, 000 male births. The higher incidence of hemophilia A may be due to the greater amount of DNA “at risk” for mutation in the factor VIII gene (186, 000 base pairs) compared with the factor IX gene (34, 000 base pairs). Hemophilia A and B are observed throughout all races and ethnic groups, and more than 25, 000 individuals in the United States are recognized to have one of the hemophilias. Although carrier testing, genetic counseling, and prenatal diagnosis are widely available in the United States through the network of federally funded Hemophilia Treatment Centers, fecundity rates remain high, and few confirmed carriers elect to terminate their pregnancies even if an affected fetus is detected in utero. These decisions probably are influenced by the wide availability of efficacious and safe commercial coagulation factor replacement concentrates and by the prospect that gene therapy may eventually cure the hemophilias
• Genetics • As with other sex-linked recessive diseases, the genes for factor VIII and factor IX are located on the long arm of the X chromosome. Males with a defective allele on their single X chromosome do not transmit this gene to their sons, but all of their daughters become obligate carriers. Female carriers transmit the coagulation disorder to half of their sons, and half of their daughters become carriers. Female carriers can manifest hemophilia-like symptoms if the alleles on the X chromosome are unequally inactivated (lyonization); the defective hemophilic allele is expressed in preference to the normal allele, and a phenotypic hemophiliac is produced. Female hemophilia can arise as the result of mating between a hemophilic male and a female carrier (homozygous for the defective factor VIII or IX gene) or in carrier females who have the 45 XO karyotype (Turner's syndrome) and are hemizygous for the defective hemophilia gene
• Clinical Manifestations • The clinical pictures of hemophilia A and hemophilia B are indistinguishable from each other, with their clinical severity corresponding inversely to the circulating levels of plasma coagulant factor VIII or IX activity. Individuals with less than 1% of normal factor VIII or IX activity have severe disease characterized by frequent spontaneous bleeding events in joints (hemarthrosis) and soft tissues and by profuse hemorrhage with trauma or surgery. Spontaneous bleeds are uncommon with mild deficiencies (>5% normal activity); however, excessive bleeding still can occur with trauma or surgery. A moderate clinical course is associated with factor VIII or IX levels between 1 and 5%. Approximately 60% of all cases of hemophilia A are clinically severe, whereas only 20 to 45% of cases of hemophilia B are severe
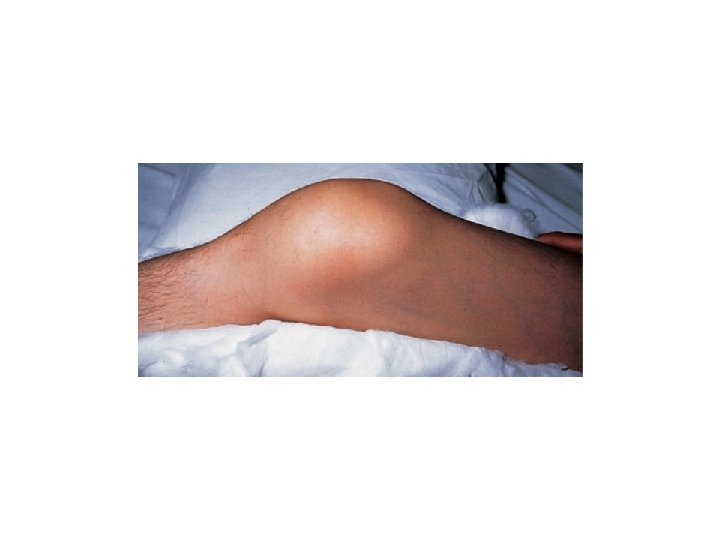
• Severe hemophilia typically is suspected and diagnosed during infancy in the absence of a family history. • Although the trauma of uncomplicated childbirth (vaginal or cesarean section) rarely produces intracranial hemorrhage, prolonged labor, forceps delivery, and the use of vacuum extraction are major risk factors. • Circumcision within days after birth is accompanied by excessive bleeding in fewer than half of severely affected boys. • The first spontaneous hemarthrosis in severely affected hemophiliacs usually occurs between 12 and 18 months of age, when ambulation begins, and in moderately affected individuals it occurs at about 2 to 5 years of age. • The knees are the most prominent sites of spontaneous bleeds, followed by the elbows, ankles, shoulders, and hips; wrists are less commonly involved
• Recurrent or untreated bleeds result in chronic synovial hypertrophy and eventually damage to the underlying cartilage, with subsequent subchondral bone cyst formation, bony erosion, and flexion contractures. The intra-articular iron deposited in hemarthroses may activate oncogenes, which subsequently stimulate synovial proliferation. Abnormal mechanical forces from weight bearing can produce subluxation, misalignment, loss of mobility, and permanent deformities of the lower extremities (). These changes are accompanied by chronic pain, swelling, and arthritis. Plain radiographs and clinical examination of chronic hemarthroses often underestimate the extent of bone and joint damage; serial magnetic resonance imaging (MRI) appears to be the most sensitive and specific means of detecting and monitoring early and progressive disease
• Diagnosis • The diagnosis of hemophilia in infancy is confirmed by detection of significantly reduced factor VIII or IX activities in the plasma of male babies born into families known to be affected by hemophilia or of male children who present with excessive bruising or bleeding at the time of circumcision, when intramuscular injections of immunizing vaccinations are administered, or after trauma during the toddler years. Hemarthrosis, the most common cause of morbidity in severe hemophilia, is not prevalent in pretoddler years. The diagnosis of mild hemophilia B in the neonate may be confounded by the fact that factor IX activity may be decreased substantially in normal infants owing to reduced hepatic synthesis of vitamin K–dependent proteins by an immature liver
• Newborns without hemophilia have reduced levels of factor IX (to approximately 40% of normal), and there is a gradual rise during the first year of life into the low-normal adult range. Prematurity is associated with even lower factor IX levels due to the immaturity of the liver. Evaluation of suspected hemophilia in a female should exclude von Willebrand's disease (v. WD) and its variants (e. g. , type 2 Normandy), as well as the rare occurrence of a normal male karyotype associated with testicular feminization. Molecular genetic testing may be useful to confirm suspected hemophilia in any patient with low circulating levels of factor VIII or factor IX
• Treatment • Reversal and prevention of acute bleeding events in hemophilia A and B are based on replacement of the missing or deficient clotting factor protein to restore adequate hemostasis. The morbidity, mortality, and overall cost of care for individuals with hemophilia are reduced significantly if care is provided by comprehensive hemophilia centers, where the multispecialty expertise, specialized coagulation laboratory, and diagnostic capabilities exist to coordinate and monitor specific patient needs.
• Replacement guidelines are intended to achieve plasma levels of factor VIII and IX activity of 25 to 30% for minor spontaneous or traumatic bleeds (e. g. , hemarthroses, persistent hematuria), at least 50% clotting factor activity for the treatment or prevention of severe bleeds (e. g. , major dental surgery, maintenance replacement therapy after major surgery or trauma), and 80 to 100% activity for any lifethreatening or limb-threatening hemorrhagic event (e. g. , major surgery, trauma). After major trauma or if visceral or intracranial bleeding is suspected, replacement therapy adequate to achieve 100% clotting factor activity should be administered before diagnostic procedures are initiated. Although replacement dosing is often empiric, plasma factor VIII activity increases about 2% (0. 02 IU/m. L) for each unit of factor VIII administered per kilogram of body weight, and factor IX activity increases about 1% (0. 01 IU/m. L) for each unit of factor IX administered per kilogram of body weight.
• The initial dose of factor IX diffuses into the extravascular space and binds to endothelial cell surfaces to a much greater degree than is observed with factor VIII. A 70 -kg individual with severe hemophilia A or B (factor VIII or IX activity <1% of normal) who requires replacement to 100% activity for major surgery initially should receive 3500 IU of factor VIII or 7000 IU of factor IX concentrate. The circulating kinetics of factors VIII and IX require subsequent dosing every 8 to 12 hours and every 18 to 24 hours, respectively, and dosing should be individualized according to the peak recovery increment within 15 to 30 minutes after bolus infusion as well as trough activity levels. The frequency of repeat dosing also is determined by the rapidity of pain relief, recovery of joint function, and resolution of active bleeding. Replacement usually is maintained for 10 to 14 days after major surgery to allow for proper wound healing.
• Bolus dosing typically results in wide fluctuations in clotting factor activity levels and requires frequent laboratory monitoring to avoid suboptimal troughs. Continuous infusion regimens, consisting of 1 to 2 IU of factor VIII or IX concentrate per kilogram per hour after a bolus dose, maintain a plateau level without the necessity for frequent laboratory testing and reduce total concentrate consumption by 30 to 75% in surgical settings.
• Cryoprecipitate (the precipitate remaining after fresh-frozen plasma [FFP] is thawed at 4° C) and FFP contain factor VIII, but factor IX is contained only in FFP. However, they are not the optimal replacement products for either hemophilia A or hemophilia B because of their potential to transmit blood-borne pathogens. Plasma-derived clotting factor concentrates are manufactured from the plasma donations pooled from thousands of individual donors and are subjected to various types of viral inactivation techniques. Only lipid-enveloped viruses are susceptible to these procedures, which increases the risk that these products can transmit viruses such as parvovirus B 19, hepatitis A, and prions, which have been implicated in variant Creutzfeldt-Jakob disease (v. CJD). The safety of these products has been enhanced by deferring the inclusion of firsttime donor plasma collections from the plasma pool and by implementing more rigorous and specific viral surveillance of “minipools” (16 individual plasma donors) prior to manufacture