Chemistry of Aromatic Compounds Department of Pharmaceutical Analysis


 is intermediate between")





, based on quantum mechanics: • Any conjugated monocyclic polyene that is planar")






















 C—Cl C—Br CH 3—X 1. 77 1. 91 C 2 H")


 Grignard reagent")
 EAS The –X group is electron-withdrawing and deactivating in EAS, but is an")
 Nucleophilic aromatic substitution (bimolecular displacement) Ar must contain strongly electron withdrawing groups ortho")
 mechanism:")

 Elimination-Addition, nucleophilic aromatic substitution. When the ring is not activated to the bimolecular")
")




 Basicity of Amines. The lone pair of electrons on nitrogen")
NH 3+ 10. 8")
 make the substituted aniline more basic than")








3 CCH 2 CNH")

")








 nitration")
 sulfonation")
 halogenation")

 Friedel-Crafts alkylation NR with –NH 2, -NHR, -NR 2")
 Friedel-Crafts acylation NR with –NH 2, -NHR, -NR 2")
 nitrosation")
 coupling with diazonium salts azo dyes")

 Reacts to produce a clear")


 is prepared in situ")

 salts Advantages of")








")



: HBase")




")
 Ar-O-Na+ + R-X Ar-O-R + Na. X note: R-X")

 halogenation")
 sulfonation At low temperature the reaction is non-reversible and the lower Eact ortho")
 Friedel-Crafts alkylation.")
 Friedel-Crafts acylation")

 nitrosation")
 coupling with diazonium salts (EAS with the weak electrophile diazonium)")
 Kolbe reaction (carbonation)")
 Reimer-Tiemann reaction")



 d c b a")

- Slides: 124
Chemistry of Aromatic Compounds Department of Pharmaceutical Analysis Chalapathi Institute of Pharmaceutical Sciences
The Structure of Benzene • Because each bond has two electrons, benzene has six electrons.
• In benzene, the actual bond length (1. 39 Å) is intermediate between the carbon—carbon single bond (1. 53 Å) and the carbon—carbon double bond (1. 34 Å).
Spectroscopic Properties of Benzene
• The resonance description of benzene consists of two equivalent Lewis structures, each with three double bonds that alternate with three single bonds. • The true structure of benzene is a resonance hybrid of the two Lewis structures, with the dashed lines of the hybrid indicating the position of the bonds. • We will use one of the two Lewis structures and not the hybrid in drawing benzene. This will make it easier to keep track of the electron pairs in the bonds (the electrons).
Stability of Benzene • Benzene reacts slowly with Br 2 to give bromobenzene (where Br replaces H) • This is a substitution reaction rather than the rapid addition reaction common to compounds with C=C.
Heats of Hydrogenation as Indicators of Stability • The addition of H 2 to C=C normally gives off about 118 k. J/mol – 3 isolated double bonds would give off 356 k. J/mol – Two conjugated double bonds in cyclohexadiene add 2 H 2 to release 230 k. J/mol • Benzene has 3 units of unsaturation but gives off only 206 k. J/mol on reacting with 3 H 2 molecules • Therefore it has about 150 k. J more “stability” than an isolated set of three double bonds
n. Benzene is actually 24 k. J more stable than cyclohexadiene!
Hückel’s Rule(1931), based on quantum mechanics: • Any conjugated monocyclic polyene that is planar and has (4 n+2)π and/or nonbonding electrons, with n = 0, 1, 2, etc. , will exhibit the special stability associated with aromaticity. (1930) • n 0 1 2 3 4 4 n + 2 Pi electrons 4(0) + 2 = 2 4(1) + 2 = 6 4(2) + 2 = 10 4(3) + 2 = 14 4(4) + 2 = 18
Which are Aromatic? ? ?
Electrophilic Aromatic Substitution
Nitration
Nitration Mechanism
Sulfonation is Reversible
Desulfonation
Bromination / Chlorination
Bromination Mechanism
Reaction Profile
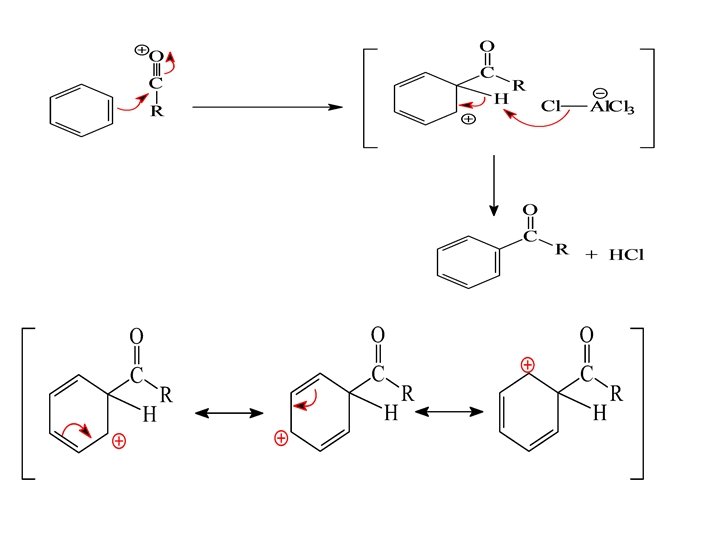
Friedel-Crafts Acylation
Friedel-Crafts Alkylation
Mechanism of Friedel-Crafts Alkylation
Bisubstitutions, directing Effects
Substituent Summary
Activating group Summary
Electron-Withdrawing Nitro Group effect
Deactivating group summary
Halogens are the Anomoly Deactivators and o, p-Directors By electron withdrawing inductive effect, halogens deactivates the rings. Resonance donation causes o, p direction
Di substitution reactions Nitration of toluene Acylation of p-methyl toluene
Reactions of rings with 2 or more substituents
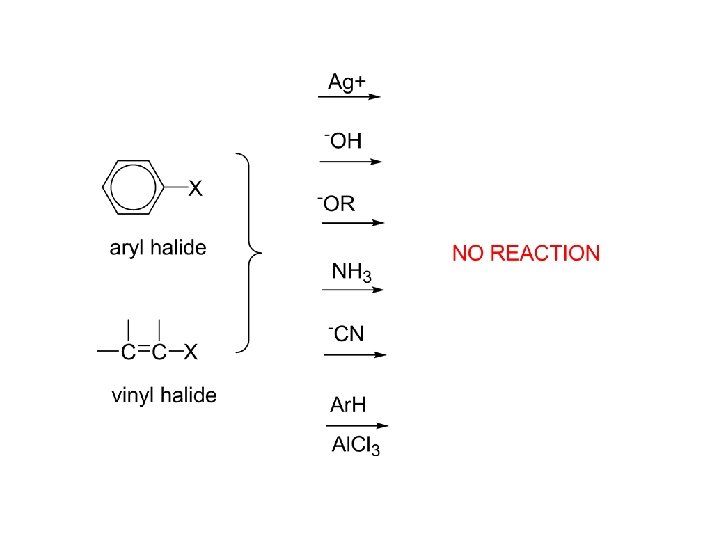
Aryl Halides Ar-X Organic compounds with a halogen atom attached to an aromatic carbon are very different from those compounds where the halogen is attached to an aliphatic compound. Carbon-halogen bonds in aryl halides are shorter and stronger than carbon-halogen bonds in alkyl halides. Because the carbonhalogen bond is stronger, aryl halides react more slowly than alkyl halides when carbon-halogen bond breaking is rate determining. While the aliphatic compounds readily undergo nucleophilic substitution and elimination reactions, the aromatic compounds resist nucleophilic substitution, only reacting under severe conditions or when strongly electron withdrawing groups are present ortho/para to the halogen.
reactions of alkyl halides Ar-X 1. SN 2 NR 2. E 2 3. organo metallic compounds 4. reduction NR similar
Bond Lengths (Å) C—Cl C—Br CH 3—X 1. 77 1. 91 C 2 H 5—X 1. 77 1. 91 sp 3 (CH 3)3 C—X 1. 80 1. 92 CH 2=CH—X 1. 69 1. 86 C 6 H 5—X 1. 69 1. 86 sp 2
In aryl halides, the carbon to which the halogen is attached is sp 2 hybrizided. The bond is stronger and shorter than the carbon-halogen bond in aliphatic compounds where the carbon is sp 3 hybridized. Hence it is more difficult to break this bond aryl halides resist the typical nucleophilic substitution reactions of alkyl halides. The same is true of vinyl halides where the carbon is also sp 2 hybridized and not prone to nucleophilic substitution. In a manner analogous to the phenols & alcohols, we have the same functional group in the two families, aryl halides and alkyl halides, but very different chemistries.
Aryl halides, reactions: 1. Formation of Grignard reagent 2. EAS 3. Nucleophilic aromatic substitution (bimolecular displacement) (Ar must contain strongly electron withdrawing groups ortho and/or para to X) 4. Nucleophilic aromatic substitution (elimination-addition) (Ring not activated to bimolecular displacement)
1) Grignard reagent
2) EAS The –X group is electron-withdrawing and deactivating in EAS, but is an ortho/para director.
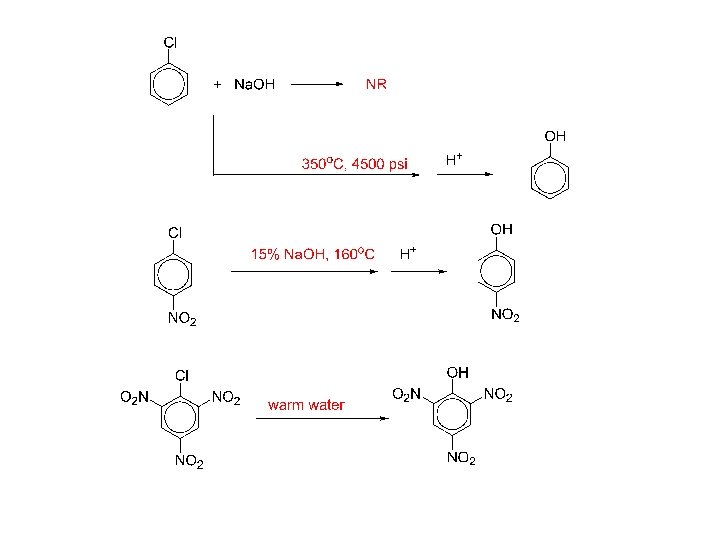
3) Nucleophilic aromatic substitution (bimolecular displacement) Ar must contain strongly electron withdrawing groups ortho and/or para to the X.
bimolecular displacement (nucleophilic aromatic substitution) mechanism:
evidence for the bimolecular displacement mechanism: no element effect : Ar-I Ar-Br Ar-Cl Ar-F (the C—X bond is not broken in the RDS)
4) Elimination-Addition, nucleophilic aromatic substitution. When the ring is not activated to the bimolecular displacement and the nucleophile is an extremely good one.
Elimination-Addition mechanism (nucleophilic aromatic substitution)
While the concept of “benzyne” may appear to be strange, there is much evidence that this mechanism is correct.
benzyne intermediate has been trapped in a Diels-Alder condensation:
Amines. Organic derivatives of ammonia, NH 3. Nitrogen atom have a lone pair of electrons, making the amine both basic and nucleophilic Amines Nomenclature. (please read) alkylamines arylamines Amines are classified according to the degree of nitrogen substitution: 1° (RNH 2), 2° (R 2 NH), 3° (R 3 N) and 4° (R 4 N+) primary (1°) amines secondary (2°) amines tertiary (3°) amines quarternary (4°) ammonium ion Note: Although the terminology is the same, this classification of amines is different from that of alcohols. 48
Structure and bonding. The nitrogen of alkylamines is sp 3 hybridized and tetrahedral. The nitrogen of arylamines (aniline) is slightly flatten, reflecting resonance interactions with the aromatic ring.
Physical Properties. (please read) Basicity of Amines. The lone pair of electrons on nitrogen makes amines basic and nucleophilic. They react with acids to form acid–base salts and they react with electrophiles The basicity is reflective of and is expressed as the p. Ka of the conjugate acid. The conjugate base of a weak acid is a strong base: Higher p. Ka = weaker acid = stronger conjugate base The conjugate base of a strong acid is a weak base Lower p. Ka = stronger acid = weaker conjugate base p. Ka values of ammonium ions Alkyl ammonium ions, R 3 NH+ X-, have p. Ka values in the range of 10 -11 (ammonium ion, H 4 N+ X-, has a p. Ka ~ 9. 3) The ammonium ions of aryl amines and heterocyclic aromatic amines are considerably more acidic than alkyl amines (p. Ka < 5). The nitrogen lone pair is less basic if it is in an sp 2 hybridized orbital (versus an sp 3)
NH 4+ p. Ka= 9. 3 (H 3 CH 2 C)NH 3+ 10. 8 (H 3 CH 2 C)2 NH 2+ 11. 1 (H 3 CH 2 C)3 NH+ 10. 8 p. Ka= 4. 6 5. 2 0. 4 7. 0 - 1. 0 Arylamines are much less basic than alkylamines. The lone pair of electrons on the nitrogen of aniline are conjugated to the -electrons of the aromatic ring and are therefore less available for acid-base chemistry. Protonation disrupts the conjugation. Substitutents can greatly influence the basicity of the aniline. The effect is dependent upon the nature and position of the substitutent.
Electron-donating substituents (-CH 3, -OH, -OCH 3) make the substituted aniline more basic than aniline itself (the p. Ka of the anilinium ion is higher than 4. 6) Electron-withdrawing substituents (-Cl, -NO 2) make the substituted aniline less basic than aniline itself (the p. Ka of the anilinium ion is lower than 4. 6)
Y= -NH 2 -OCH 3 p. Ka= 5. 3 -CH 3 -H -Cl -CF 3 -CN -NO 2 p. Ka= 6. 2 p. Ka= 5. 1 p. Ka= 4. 6 p. Ka= 4. 0 p. Ka= 3. 5 p. Ka= 1. 7 p. Ka= 1. 0 less acidic (more basic) more acidic (less basic)
Preparation of amines 1. Alkylation or ammonolysis of alkyl halides: Ammonia and other alkylamines are good nucleophiles and react with 1° and 2° alkyl halides or tosylates via an SN 2 reaction yielding alkyl amines. 1°, 2°, and 3° amines all have similar reactivity; the initially formed monoalkylation product can undergo further reaction to yield a mixture of alkylated products
2. Reduction of Alkyl azides, nitriles, amides, and nitroarene can be reduced to the corresponding amines. Li. Al. H 4 reduces azides to 1° amines Li. Al. H 4 reduces nitriles to 1° amines Nitroarenes are reduced to anilines Li. Al. H 4 reduces amides to 1°, 2° or 3° amines
3. Reductive Amination. easily reduced to amines. Imines and iminium ions are
Sodium cyanoborohydride, Na+ N C-BH 3– : the cyano ligand makes cyanoborohydride a weak hydride source and it will react with only the most easily reduced functional groups, such as an iminium ion. Na. B(CN)H 3 reduces ketones and aldehydes slowly.
4. Hofmann and Curtius rearrangements • Carboxylic acid derivatives can be converted into primary amines with loss of one carbon atom by both the Hofmann rearrangement and the Curtius rearrangement
Curtius reaction mechanism • Heating an acyl azide prepared from an acid chloride • Migration of R from C=O to the neighboring nitrogen with simultaneous loss of a leaving group
Hofmann Rearrangement • RCONH 2 reacts with Br 2 and base • Gives high yields of arylamines and alkylamines • Figure 24. 5 See Mechanism O Br 2 RCNH 2 HO– R N H 2 + CO 32–
Examples of hofmann reaction O Br 2, Na. OH (CH 3)3 CCH 2 CNH 2 (CH 3)3 CCH 2 NH 2 H 2 O (94%) O Br 2, KOH CNH 2 H 2 O Br Br (87%)
Mechanism of the Hofmann Rearrangement O O RCNH 2 RCNHBr RNCO R N H 2 • The Hofmann rearrangement involves 6 steps in 3 stages. • 1. formation of an N-bromo amide (2 steps) • 2. conversion of the N-bromo amide to an isocyanate (2 steps) • 3. hydrolysis of the isocyanate (2 steps)
5. Alkylation (ammonolysis of alkyl halides)
Reactions of Amines 1. Alkylation and acylation
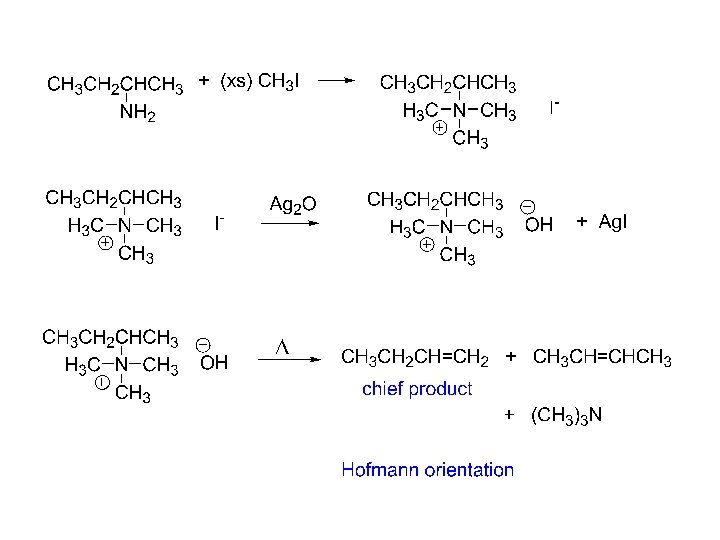
2. Hofmann Elimination • Converts amines into alkenes • NH 2 is very a poor leaving group so it converted to an alkylammonium ion, which is a good leaving group
Orientation in Hofmann Elimination • We would expect that the more highly substituted alkene product predominates in the E 2 reaction of an alkyl halide (Zaitsev's rule) • However, the less highly substituted alkene predominates in the Hofmann elimination due to the large size of the trialkylamine leaving group • The base must abstract a hydrogen from the most sterically accessible, least hindered position
Steric Effects Control the Orientation
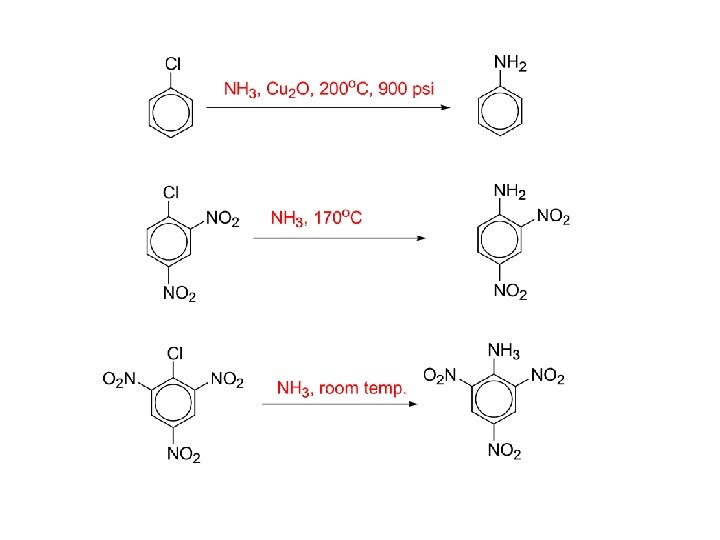
3. The Sandmeyer Reaction: Primary arylamines react with HNO 2, yielding stable arenediazonium salts. The N 2 group can be replaced by a nucleophile. Reaction of an arenediazonium salt with Cu. Cl or Cu. Br gives aryl halides (Sandmeyer Reaction) Aryl iodides form from reaction with Na. I without a copper(I) salt
An arenediazonium salt and Cu. CN yield the nitrile, Ar. CN, which can be hydrolyzed to Ar. COOH From reaction of the arenediazonium salt with copper(I) oxide in an aqueous solution of copper(II) nitrate
• By treatment of a diazonium salt with hypophosphorous acid, H 3 PO 2
5. EAS -NH 2, -NHR, -NR 2 are powerful activating groups and ortho/para directors a) nitration b) sulfonation c) halogenation d) Friedel-Crafts alkylation e) Friedel-Crafts acylation f) coupling with diazonium salts g) nitrosation
a) nitration
b) sulfonation
c) halogenation
Swimming pool test kit for chlorine:
e) Friedel-Crafts alkylation NR with –NH 2, -NHR, -NR 2
f) Friedel-Crafts acylation NR with –NH 2, -NHR, -NR 2
g) nitrosation
h) coupling with diazonium salts azo dyes
6. Hofmann elimination from quarternary hydroxides step 1, exhaustive methylation 4 o salt step 2, reaction with Ag 2 O 4 o hydroxide + Ag. X step 3, heat to eliminate alkene(s) + R 3 N
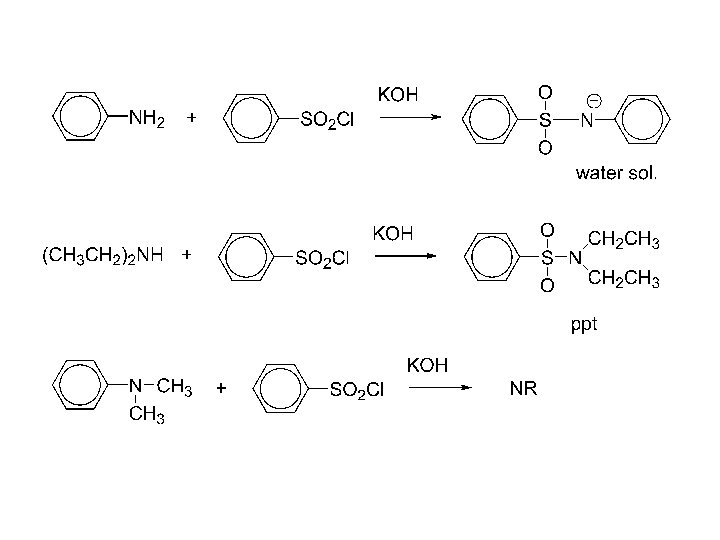
Hinsberg Test: unknown amine + benzenesulfonyl chloride, KOH (aq) Reacts to produce a clear solution and then gives a ppt upon acidification primary amine. Reacts to produce a ppt secondary amine. Doesn’t react tertiary amine.
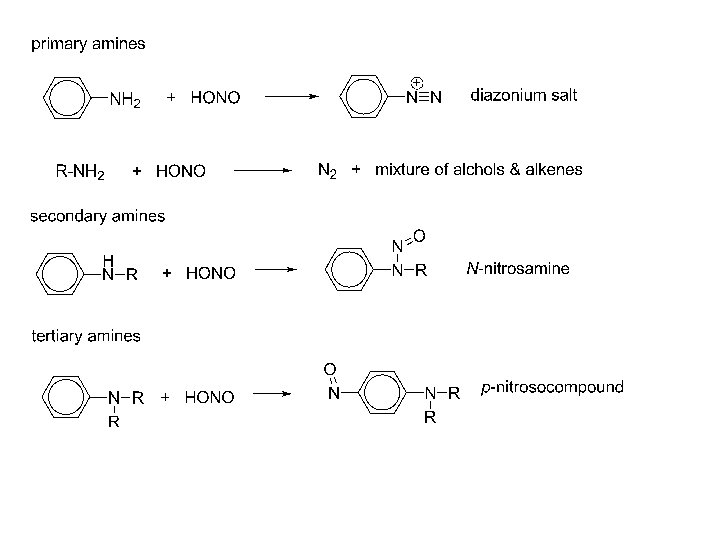
Diazonium Salts Diazonium: there are 2 nitrogen atoms joined together in the positive ion. In French, nitrogen is still called by its old name ‘azote’ which means unable to support life. Notice the triple bond between the nitrogen atoms The positive charge is on the nitrogen that is attached to the benzene ring. They are essential in the dye industry. A Diazonium salt is produced then reacted with a phenol. If the correct phenol is used, almost any colour can be produced.
Formation of the Diazonium salt: Formed by reacting phenylamine with sodium nitrite and hydrochloric acid. These reagents form in situ nitrous acid HONO. The Diazonium salt is unstable above 10°C, so the reaction is normally carried out in ice. An aliphatic Diazonium salt is very unstable, so only aromatics are used. The lone pairs present in the salt can participate in the benzene ring, making it more stable. More correctly this is due to overlap of p-orbitals in the diazo group with the p-system in the ring. So phenylamine would give benzenediazonium chloride.
The conditions are 5°C and remember the HONO (nitrous acid) is prepared in situ by reacting sodium nitrite with hydrochloric acid. The diazonium salt can ten do one of two things depending on the temperature
Hydrolysis coupling
Synthetic Transformations of Aryl Diazonium Salts. Sandmeyer reaction: promoted by Cu(I) salts Advantages of the aryl diazonium salt intermediate: 1) Introduces aryl substituents that are not otherwise accessible, such as -OH, -F, -I, and -CN. 2) Allows preparation of substituted arenes with substitution patterns that can not be prepared by other means.
Synthesis 3, 5 -dibromotoluene
REACTIONS OF PHENOLS AND DIAZONIUM SALTS
Phenols Ar-OH Phenols are compounds with an –OH group attached to an aromatic carbon. Although they share the same functional group with alcohols, where the –OH group is attached to an aliphatic carbon, the chemistry of phenols is very different from that of alcohols.
Nomenclature. Phenols are usually named as substituted phenols. The methylphenols are given the special name, cresols. Some other phenols are named as hydroxy compounds.
physical properties phenols are polar and can hydrogen bond phenols are water insoluble phenols are stronger acids than water and will dissolve in 5% Na. OH phenols are weaker acids than carbonic acid and do not dissolve in 5% Na. HCO 3
Intramolecular hydrogen bonding is possible in some ortho-substituted phenols. This intramolecular hydrogen bonding reduces water solubility and increases volatility. Thus, o-nitrophenol is steam distillable while the isomeric p-nitrophenol is not.
phenols, syntheses: 1. From diazonium salts 2. Alkali fusion of sulfonates
Reactions: alcohols phenols 1. HX NR 2. PX 3 NR 3. dehydration NR 4. as acids phenols are more acidic 5. ester formation similar 6. oxidation NR
Phenols, reactions: 1. as acids 2. ester formation 3. ether formation 4. EAS a) nitration f) nitrosation b) sulfonation g) coupling with diaz. salts c) halogenation d) Friedel-Crafts alkylation e) Friedel-Crafts acylation h) Kolbe i) Reimer-Tiemann
as acids: with active metals: with bases: CH 4 < NH 3 < HC CH < ROH < H 2 O < phenols < H 2 CO 3 < RCOOH < HF
CH 4 < NH 3 < HC CH < ROH < H 2 O < phenols < H 2 CO 3 < RCOOH < HF
CH 4 < NH 3 < HC CH < ROH < H 2 O < phenols < H 2 CO 3 < RCOOH < HF water phenols 5% Na. OH 5% Na. HCO 3 insoluble soluble carboxylic acids
We use the ionization of acids in water to measure acid strength (Ka): HBase + H 2 O H 3 O+ + Base- Ka = [H 3 O+ ][ Base ] / [ HBase] ROH Ka ~ 10 -16 - 10 -18 Ar. OH Ka ~ 10 -10 Why are phenols more acidic than alcohols?
ROH + H 2 O H 3 O+ + RO- Ar. OH + H 2 O H 3 O+ + Ar. O- Resonance stabilization of the phenoxide ion, lowers the PE of the products of the ionization, decreases the ΔH, shifts the equil farther to the right, makes phenol more acidic than an alcohol
effect of substituent groups on acid strength? Electron withdrawing groups will decrease the negative charge in the phenoxide, lowering the PE, decreasing the ΔH, shifting the equil farther to the right, stronger acid. Electron donating groups will increase the negative charge in the phenoxide, increasing the PE, increasing the ΔH, shifting the equilibrium to the left, weaker acid.
Number the following acids in decreasing order of acid strength (let # 1 = most acidic, etc. ) 3 5 1 4 2
1 2 3 4
2. ester formation (similar to alcohols)
3. ether formation (Williamson Synthesis) Ar-O-Na+ + R-X Ar-O-R + Na. X note: R-X must be 1 o or CH 3 Because phenols are more acidic than water, it is possible to generate the phenoxide in situ using Na. OH.
4. Electrophilic Aromatic Substitution The –OH group is a powerful activating group in EAS and an ortho/para director. a) nitration
b) halogenation
c) sulfonation At low temperature the reaction is non-reversible and the lower Eact ortho -product is formed (rate control). At high temperature the reaction is reversible and the more stable paraproduct is formed (kinetic control).
d) Friedel-Crafts alkylation.
e) Friedel-Crafts acylation
Fries rearrangement of phenolic esters.
f) nitrosation
g) coupling with diazonium salts (EAS with the weak electrophile diazonium)
h) Kolbe reaction (carbonation)
i) Reimer-Tiemann reaction
Spectroscopy of phenols: Infrared: O—H stretching, strong, broad 3200 -3600 cm-1 C—O stretch, strong, broad ~1230 cm-1 (alcohols ~ 1050 – 1200) nmr: O—H 4 -7 ppm (6 -12 ppm if intramolecular hydrogen bonding)
o-cresol C--O O--H
o-cresol c b a
ethyl salicylate (intramolecular hydrogen bonding) d c b a
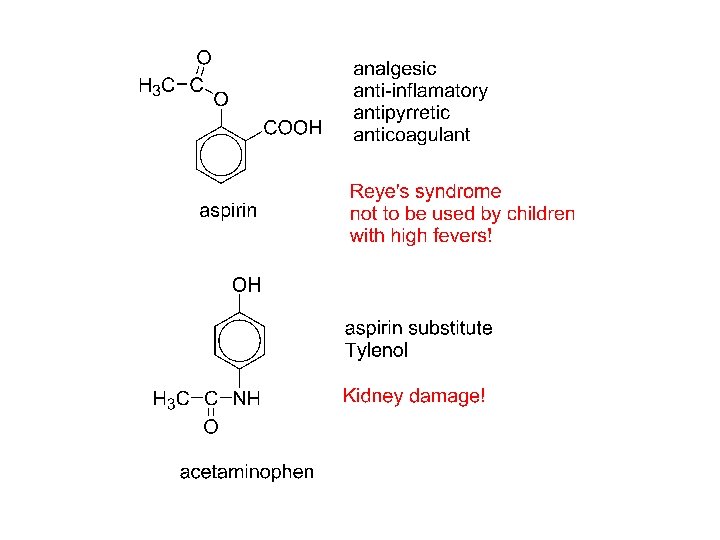
Thank you