Pharmacologie et thrapeutiques UE 2 11 IFSI 1re


 - Absorption / résorption - Distribution - Métabolisation")





, le PA doit être")




















 �Certains médicaments ont une affinité")

 •")






 Glomérule Artériole afférente 1 Artériole efférente NEPHRON 2 3 Urine")
 de la fraction libre. Réabsorption de molécule")











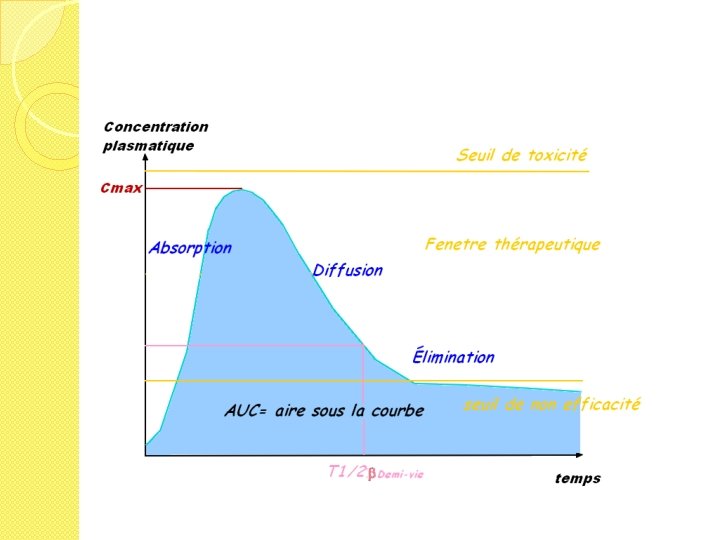








- Slides: 60
Pharmacologie et thérapeutiques UE 2. 11 IFSI 1ère année Pharmacocinétique F. Bengeloun – dec. 2014
Définition � Étudie le sort du médicament dans l’organisme, depuis son administration jusqu’à son élimination � Permet de choisir la voie d ’administration et d ’adapter la posologie � Permet d’établir des règles d’évolution des concentrations du médicament en fonction du temps � Rôle : application de ces règles à un malade donné de façon à ajuster son traitement en fonction des paramètres qui lui sont personnels (âge, poids, pathologies associées…)
Définition On distingue 4 phases (ADME) - Absorption / résorption - Distribution - Métabolisation - Excrétion
Devenir du médicament dans l’organisme
Devenir du médicament dans l’organisme
PA dans une forme pharmaceutique solide Ex comprimé Désintégration Libération PA en particules Dissolution Absorption PA en solution Ex solution injectable PA dans les tissus Distribution PA dans le sang Récepteur Elimination Phase Biopharmaceutique PA éliminé Réponse Pharmacocinétique Phase Pharmacodynamique
Phase biopharmaceutique
Libération • 1ère étape de la mise à disposition du PA après l’administration extravasculaire d’une forme pharmaceutique solide • Elle peut se faire o Rapidement dans le cas d’une forme pharmaceutique à libération rapide o Lentement dans le cas d’une forme à libération prolongée o De manière retardée dans le cas d’une forme à libération retardée
Dissolution • Pour pouvoir traverser les membranes (ex muqueuse digestive), le PA doit être à l ’état de molécules dissoutes • La vitesse de dissolution dépend des caractéristiques o du PA o et aussi du site de l’absorption
Phase pharmacocinétique
Devenir du médicament dans l’organisme = Pharmacodynamie
Absorption / Résorption �Pénétration du médicament dans le sang à partir de son lieu d’absorption �Le PA dissous traverse les membranes biologiques pour pénétrer dans le circulation sanguine �Cette étape n’existe pas lorsque le médicament est introduit directement dans la circulation par voie intraveineuse �Absorption / résorption en fonction des différentes voies d’administration
Facteurs pouvant modifier la résorption • Facteurs physiopathologiques l’âge, l’activité physique, la grossesse, la vitesse de vidange gastrique, le p. H digestif, la diarrhée ou la constipation, certaines maladies chroniques, … • Facteurs exogènes o L’alimentation ( ex : modification de la vitesse et/ou de l’intensité de la résorption - Laitage, jus de pamplemousse…) o Médicaments associés interactions médicamenteuses (ex : Maalox + Ciprofloxacine. Les sels de Mg du Maalox précipitent avec la Ciprofloxacine => pas de résorption de l’antiobiotique)
Facteurs pouvant modifier la résorption • Les caractéristiques du médicament o Physico-chimiques : p. Ka o Hydro / lipo solubilité o Taille et forme de la molécule o Forme galénique (sirop, comprimé, gélule…), qui détermine la vitesse de la phase biopharmaceutique
Absorption / Résorption � Quelle que soit la voie d’administration, le passage des membranes biologiques, du site d’administration vers un compartiment sanguin s’effectue selon deux mécanismes principaux : ◦ Diffusion passive = selon lipophilie du PA, la taille de la molécule, son gradient de concentration de part et d’autre de la membrane ◦ Transport actif (avec transporteur)
Absorption / Résorption
Notion de biodisponibilité = Evaluation de l’absorption �Fraction de la dose administrée qui atteint la circulation générale �Elle dépend : ◦ De la dégradation du PA (tube digestif, tissus musculaire, pulmonaire…) ◦ De l’importance de sa résorption ◦ De l’existence d’un effet de 1 er passage hépatique �Biodisponibilité par voie IV = 100 %
Notion de biodisponibilité �Faible biodisponibilité n’équivaut pas à mauvaise activité �La captation hépatique peut générer des métabolites actifs �Une faible biodisponibilité peut être compensée par un dosage plus élevé �Une faible biodisponibilité est par contre plus sujette à des variations intra- individuelles car moins constante
Absorption : Notion de 1 er passage hépatique �En fonction du lieu de résorption, le médicament peut être conduit directement via la veine porte dans le tissus hépatique. A ce niveau, il est plus ou moins dégradé avant même d’avoir pu être distribué dans l’organisme �Administration sans 1 er passage hépatique : sublinguale, inhalée et transdermique. �http: //www. icp. org. nz/icp_t 6. html
Absorption : Notion de 1 er passage hépatique Lors de la prise orale d'un médicament, suivie d'une absorption gastrointestinale, la substance active est d'abord transportée par la veine porte vers le foie avant d'atteindre la circulation systémique
Devenir du médicament dans l’organisme = Pharmacodynamie
Distribution • Après l’absorption, le PA parvient dans le plasma où il se trouve sous deux formes : ◦ Forme liée aux protéines plasmatiques – Sorte de réserve en PA ( inactivation temporaire du médicament) ◦ Forme libre (en solution dans le plasma) seule responsable de l’action pharmacologique PA + Prot PAProt � Le médicament se fixe sur les protéines plasmatiques (surtt albumine) qui le transportent � Fixation aux protéines plasmatiques variable et réversible
Fixation protéique d’après E. Singlas, A; M. Taburet Type 1 Type 2 Nature du médicament Acide faible Base faible et susbstance non ionisable Ionisation au p. H du plasma Oui/non selon nature Protéines fixatrices Albumine, lipoprotéines, globulines. . Affinité forte faible Nombre de sites (sur albumine) Petit (<4) Grand (>30) Possibilité de saturation Oui Non Risque d’interactions Oui improbable
Fixation protéique �On admet que l’action pharmacologique est proportionnelle à la concentration de la forme libre dans le plasma �Le pourcentage de fixation aux protéines est insuffisant pour comprendre les conséquences de cette fixation sur la pharmacocinétique �La fixation protéique n’est à considérer que si elle est élevée (supérieure à 90%) et si le médicament a une marge thérapeutique étroite
Facteurs modifiants la liaison aux protéines plasmatiques • Affinité du PA pour les protéines • Quantité de protéines plasmatiques Ex : antidépresseur fixation > 95 % Ex : paracétamol < 10 % • Concentration du médicament • La compétition entre 2 molécules (interactions médicamenteuses)
Facteurs modifiants la liaison aux protéines plasmatiques �Nouveaux nés: fraction libre plus élevée (et interaction avec bilirubine) � Sujets âgés: risque d’hypoalbuminémie avec fraction libre augmentée �Insuffisance hépatique et rénale: risque d’augmentation de la fraction libre par compétition avec bilirubine et métabolites divers
Diffusion tissulaire �La fraction libre du PA diffuse vers les tissus et passe du compartiment plasmatique vers le compartiment tissulaire �Le PA peut se fixer au niveau : ◦ de son site d’action effet du médicament ◦ du tissu pour lequel il a une affinité particulière (ex. les médicaments lipophiles se fixent aux tissus graisseux) risque d’effets secondaires à plus ou moins long terme.
Diffusion tissulaire � Diffusion en fonction : ◦ des propriétés physico-chimiques du médicament ◦ de l’irrigation sanguine du tissu ◦ de la perméabilité des capillaires… ◦ concept de « barrières » �hémato-encéphalique �placentaire
Diffusion tissulaire Groupe Vascularisation tissus I Très vascularisés Cœur, poumon, foie, reins, cerveau, glandes endocrines II Moins vascularisés Peau, muscles squelettiques III Peu vascularisés Tissus adipeux IV Vascularisation négligeable Os, dents, tendons, ligaments, cartilages, phanères
Diffusion tissulaire �La diffusion augmente en cas d’inflammation (méningite) �Certains médicaments ont une affinité élective pour certains tissus (tissus adipeux)
Volume distribution �Volume apparent de distribution �Volume virtuel dans lequel devrait se distribuer le médicament pour être à la même concentration que celle du plasma (en l/kg) �Renseigne sur l’intensité de la distribution �Un volume élevé signifie une forte affinité pour certains tissus ou pour tous les tissus. �http: //www. icp. org. nz/icp_t 3. html
Facteurs modifiants la distribution • Volumes liquidiens de l’organisme (âge, état de deshydratation…) • Paramètres hémodynamiques (pathologies ou troubles fonctionnels conduisant à la diminution de l’irrigation de certains organes) • Modification de la proportion de masse graisseuse • Modification des protéines plasmatiques
Devenir du médicament dans l’organisme = Pharmacodynamie
Métabolisation Les transformations métaboliques concernent la plupart des médicaments ◦ Principe : rendre le médicament plus hydrosoluble pour faciliter son élimination ◦ Site de métabolisme : foie (+++ 1 er passage hépatique), poumons, reins, intestin ◦ Réactions : oxydation, réduction, hydrolyse: enzymes dont Cytochrome P 450, conjugaison M M-OH M-O-Conjugué Élimination urinaire ou biliaire
Métabolisation � Dans certains cas le métabolisme conduit à la formation : ◦ de composés moins actifs ou inactifs ◦ de composés actifs (ex : prodrogue) ◦ de composés toxiques � Le rythme d’administration sera d’autant plus important que le métabolisme est rapide � Les variations sont importantes : - facteurs physio-pathologiques - facteurs génétiques - associations médicamenteuses
Métabolisation � Inducteurs & inhibiteurs enzymatiques : ◦ inducteurs enzymatiques : médicaments qui stimulent le métabolisme hépatique d’autres médicaments. Les médicaments qui sont co-administrés avec des inducteurs enzymatiques seront donc moins actifs. Exemple : rifampicine + oestroprogestatifs ◦ inhibiteurs enzymatiques : médicaments qui bloquent le métabolisme hépatique d’autres médicaments. Les médicaments qui sont co-administrés avec des inhibiteurs enzymatiques seront donc plus actifs. Exemple : ciclosporine et antifongique
Devenir du médicament dans l’organisme = Pharmacodynamie
Elimination �Elimination des médicaments et de leurs métabolites par la voie urinaire (la plus importante), biliaire ou pulmonaire �Excrétion par vie biliaire ou rénale sous forme inchangée �Métabolisation facilitant l’excrétion
Elimination rénale (si PM<5000) Glomérule Artériole afférente 1 Artériole efférente NEPHRON 2 3 Urine définitive 1) Filtration glomérulaire: passage de substances du sang vers l’urine 2) Réabsorption tubulaire de substances de l’urine vers le sang 3) Sécrétion finale du plasma vers l’urine
Elimination rénale �Filtration glomérulaire (pm < albumine) de la fraction libre. Réabsorption de molécule non ionisée �La filtration glomérulaire varie avec l’âge et la fonction rénale: ◦ immature chez le nouveau-né (filtration glomérulaire et sécrétion tubulaire) ◦ filtration glomérulaire diminuée chez la personne âgée
Elimination : � Biliaire = gastro-intestinale Pour les substances de haut poids moléculaire = cycle entéro-hépatique � Pulmonaire Elimination dans l’air expiré des composés volatils tels que les anesthésiques volatils, les essences ou l’alcool � Divers ◦ ◦ Lait maternel Salive Peau et phanères larmes
Elimination: variations � ge: immaturité chez préma et Nné �Induction enzymatique �Inhibition enzymatique �Facteurs génétiques �http: //www. icp. org. nz/icp_t 2. html �http: //www. icp. org. nz/icp_t 12. html
Quantification du métabolisme et de l’élimination �Notion de clairance �Notion de demi-vie plasmatique
Clairance � Clairance totale: volume de plasma totalement épuré du médicament par unité de temps � Clairance rénale: en ml/min L’élimination va tenir compte du débit de filtration glomérulaire du patient, mais aussi de la sécrétion tubulaire possible ou de la réabsorption tubulaire possible du médicament � Clairance de la créatinine , ayant un intérêt dans la mesure du débit de filtration glomérulaire, marqueur de l'activité rénale, est très fréquemment évaluée (Cockroft ou MDRD) � http: //www. icp. org. nz/icp_t 1. html
Clairance �Clairance hépatique La clairance dépend du débit sanguin hépatique et du coefficient d’extraction hépatique du médicament considéré. �Cl = Q. E (Q = débit sanguin) E = Coefficient élevé > 0, 7 Metoprolol, morphine, propranolol. . E = Coefficient faible < 0, 3 Diazepam, theophylline, valproate…
Demi vie plasmatique � Demi-vie d’élimination = T 1/2 Temps au bout duquel la concentration sanguine a diminué de moitié ( T 1/2 = 0, 693 Vd / Cl). Plus T 1/2 est courte, plus le nombre de prise sera important et la fréquence d’administration rapprochée � État d’équilibre = Steady State obtenu après administration régulière du médicament, quand les concentrations plasmatiques cessent d‘augmenter généralement au bout de 5 T 1/2 � http: //www. icp. org. nz/icp_t 4. html
Demi vie plasmatique Concentration Steady state temps � L’état d’équilibre est atteint au bout de 5 demi-vies � 99% de la dose sont éliminés en 7 demi-vies
Utilisation pratique � équilibrer l’organisme par rapport au médicament � La cinétique doit surtout être connue pour des administrations chroniques afin : ◦ d’obtenir une efficacité thérapeutique rapidement ◦ de maintenir en permanence une concentration plasmatique active ◦ d’éviter les phénomènes d’accumulation � Ces données permettent de déterminer ◦ la dose de médicament à prescrire ◦ d’évaluer la fréquence des prises Cette fréquence est évaluée de telle sorte qu’à un instant donné, la quantité de médicament injectée = quantité de médicament éliminée Etat d’équilibre
Établissement d’un schéma thérapeutique • Etablir les paramètres pharmacocinétiques du médicament après une dose unique : clairance, volume de distribution et demi-vie • Détermination de la zone thérapeutique Concentration Cmax Seuil toxique zone thérapeutique Cmin Seuil thérapeutique temps
• Établir l’intervalle de temps entre deux prises pour se maintenir dans l’intervalle thérapeutique Concentration 1 Cmax 2 Cmin 3 temps Pour définir l’intervalle, il faut connaître la demi-vie du médicament et la zone thérapeutique
• Notion de dose de charge: dose initiale pouvant être administrée lors d’un traitement afin d’avoir d’emblée une concentration efficace. On parle de BOLUS IV C Cmax Cmin t
Intérêt de la pharmacocinétique � Prévoir la concentration du médicament au niveau de son site d’action � Sa diffusion dans l’organisme � La durée de son efficacité � La transformation en métabolites et le rôle de ces derniers � Son mode d’élimination permettant de prendre certaines précautions en fonction d’états pathologiques comme l’insuffisance rénale ou hépatique.
Exemple du paracétamol � Absorption L'absorption du paracétamol par voie orale est complète et rapide. Les concentrations plasmatiques maximales sont atteintes 30 à 60 minutes après ingestion. � Distribution Le paracétamol se distribue rapidement dans tous les tissus. Les concentrations sont comparables dans le sang, la salive et le plasma. La liaison aux protéines plasmatiques est faible.
Exemple du paracétamol � Métabolisme Le paracétamol est métabolisé essentiellement au niveau du foie. Les 2 voies métaboliques majeures sont la glycuroconjugaison et la sulfoconjugaison. Cette dernière voie est rapidement saturable aux posologies supérieures aux doses thérapeutiques. Une voie mineure, catalysée par le cytochrome P 450, est la formation d'un intermédiaire réactif (le N-acétyl benzoquinone imine), qui, dans les conditions normales d'utilisation, est rapidement détoxifié par le glutathion réduit et éliminé dans les urines après conjugaison à la cystéine et à l'acide mercaptopurique. En revanche, lors d'intoxications massives, la quantité de ce métabolite toxique est augmentée.
Exemple du paracétamol � Elimination L'élimination est essentiellement urinaire. 90% de la dose ingérée est éliminée par le rein en 24 heures, principalement sous forme glycuroconjuguée (60 à 80%) et sulfoconjuguée (20 à 30%). Moins de 5% est éliminé sous forme inchangée. � La demi-vie d'élimination est d'environ 2 heures. � Variations physiopathologiques ◦ Insuffisance rénale: en cas d'insuffisance rénale sévère (clairance de la créatinine inférieure à 10 ml/min. ), l'élimination du paracétamol et de ses métabolites est retardée. ◦ Sujet âgé: la capacité de conjugaison n'est pas modifiée.
Exemple du zolpidem � Absorption Après administration orale, le zolpidem présente une biodisponibilité d'environ 70 % avec une concentration plasmatique maximale atteinte en 0, 5 à 3 heures. � Distribution Aux doses thérapeutiques, sa pharmacocinétique est linéaire. La fixation aux protéines plasmatiques est d'environ 92 %. Le volume de distribution chez l'adulte est de 0, 54 ± 0, 02 l/kg.
Exemple du zolpidem � Métabolisme et élimination Le zolpidem est éliminé sous forme de métabolites inactifs (métabolisme hépatique), principalement dans les urines (environ 60%) et les fèces (environ 40%). Il ne possède pas d'effet inducteur sur les enzymes hépatiques. � La demi-vie d'élimination plasmatique est en moyenne de 2, 4 heures (0, 7 - 3, 5 heures).
Exemple du zolpidem � Populations à risques o Chez le sujet âgé, une diminution de la clairance hépatique est observée. La concentration au pic est augmentée d'environ 50 % sans qu'il y ait d'allongement significatif de la demi-vie (3 heures en moyenne). Le volume de distribution diminue à 0, 34 ± 0, 05 l/kg o Chez les insuffisants rénaux, dialysés ou non, on observe une diminution modérée de la clairance. Les autres paramètres cinétiques ne sont pas modifiés. Le zolpidem n'est pas dialysable. o Chez les insuffisants hépatiques, la biodisponibilité du zolpidem est augmentée. Sa clairance est sensiblement réduite et la demi-vie d'élimination est allongée (environ 10 heures).
Merci pour votre attention…