Round up the usual suspects Sarcoid Idiopathic pulmonary


















 • • Connective Tissue Disease Associated ILD")
 • DAD (AIP), EP,")









 images demonstrating usual interstitial pneumonia (UIP) pattern and possible UIP")
 Scanning power microscopy showing a patchy")
 Adjacent to the regions of more chronic fibrosis (thick arrow) is a fibroblast")













- Slides: 48
Round up the usual suspects • • • Sarcoid Idiopathic pulmonary fibrosis Asbestosis, Silicosis Farmer’s lung Collagen vascular diseases Drugs Langerhans granulomatosis Lymphangitic tumor Edema More than 100 entities manifest as diffuse lung disease. Fortunately only 10 -20 of these account for about 90% of all diffuse lung disease that is assessed by open lung biopsy. De. Remee , Chest , 1987 Gurney GW: Practical approach H
IPF Classification of ILDs Am J Respir Crit Care Med 2002; 165: 279
IPF Definition • IPF is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults, limited to the lungs, and associated with the histopathologic and/or radiologic pattern of UIP • The definition of IPF requires the exclusion of other forms of interstitial pneumonia including other idiopathic interstitial pneumonias and ILD associated with environmental exposure, medication, or systemic disease
IPF Incidence and Prevalence Incidence: 10. 7 cases per 100, 000 per year for men and 7. 4 cases per 100, 000 per year for women in a population-based study from New Mexico 6. 8 and 16. 3 per 100, 000 in the United States 4. 6 per 100, 000 person-years in the United Kingdom Prevalence estimates for IPF have varied from 2 to 29 cases per 100, 000 in the general population. A recent analysis based on healthcare claims data of a large health plan in the United States yielded a prevalence estimate of between 14. 0 and 42. 7 per 100, 000 persons depending on the case definition used. It is unknown if the incidence and prevalence of IPF are influenced by geographic, ethnic, cultural, or racial factors.
Potential Risk Factors IPF Cigarette smoking Smoking is strongly associated with IPF, particularly for individuals with a smoking history of more than 20 pack-years Environmental exposures • • • metal dusts (brass, lead, steel) wood dust (pine) Farming, raising birds exposure to livestock and to vegetable dust/animal hair dressing stone cutting/polishing Microbial agents chronic viral infection in the etiology of IPF • Epstein-Barr virus , cytomegalovirus, human herpesvirus (HHV)-7, and HHV-8 • hepatitis C Gastroesophageal reflux Genetic Factors • • Familial pulmonary fibrosis (<5% of total patients with IPF): A 2 (SFTPA 2), encoding surfactant protein; ELMOD 2, a gene of unknown biological function located on chromosome 4 q 31; genetic variants within the human telomerase reverse transcriptase (h. TERT) or human telomerase RNA (h. TR) components Sporadic cases of IPF: Polymorphisms of genes encoding for cytokines (interleukin [IL]-1 a, TNFa, lymphotoxin a, IL-4, IL-6, IL 8, IL-10, and IL-12 [79– 88]), enzymes (a 1 -antitrypsin, angiotensinconverting enzyme), profibrotic molecules (TGF-b 1), coagulation pathway genes (plasminogen activator inhibitors-1 and -2), genes for surfactant protein-A and -B (70), immunomodulatory genes (complement receptor 1, NOD 2/CARD 15), and matrix metalloproteinase (MMP)-1
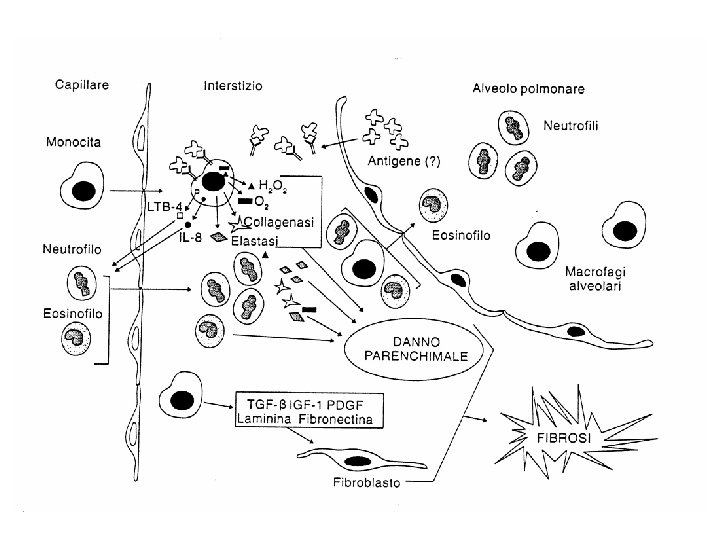
IPF Pathogenetic Hypothesis Predisposizione genetica nascita con polmone “normale” Polmone adulto esposto a stimoli “fibrogenici” Ambiente Autoimmunità tipologia di lavoro Infezioni (virali? , EBV? ) farmaci aspirazione cronica fumo Altro ? Fattori di modulazione Danno evidenziabile all’RX Manifestazione clinica
UIP/IPF DANNO Ø Infettivo? (individuati genomi di EBV ed altri Herpesvirus/antigeni virali nelle cellule epiteliali delle vie aeree di pz con IPF) Ø Autoimmune? (non trovati fin’ora nuovi autoanticorpi correlati) Ø Genetico? (predisposizione a sviluppare fibrosi dopo danno-forme familiari àAlterazione genetica nella regolazione della risposta al danno e nel metabolismo del collagene? ) Ø Neoplastico?
IPF/UIP Modelli Patogenetici 1980 2000 1990 Modelli “infiammatori” Modelli “fibrogenici”
IPF/UIP The Pathogenic Puzzle 2 Danno tissutale e riparazione 3 Attivazione fibroblastica 1 Infiammazione ed alveolite Fibrosi progressiva e rimodellamento 4 Modello classico di fibrosi infiammatoria
Contro la teoria infiammatoria: v. La flogosi non è la caratteristica istopatologica predominante nella UIP (l’alveolite sarebbe una risposta aspecifica al fumo di sigaretta, inoltre la flogosi riscontrata nella UIP è di solito lieve e raramente coinvolge aree indenni, infine altre pneumopatie dove predomina la flogosi nelle prime fasi spesso non progrediscono in fibrosi) v. Per lo sviluppo della fibrosi non è indispensabile la presenza di flogosi (esperimenti su animali transgenici hanno permesso di dissociare la risposta flogistica da quella fibrotica (IL 10 -TGFbeta Ligand) inoltre in un modello di danno epiteliale in ambiente non irrorato si è sviluppata fibrosi senza reazione infiammatoria) v. Misurazioni cliniche di flogosi non correlano con lo stadio e l’evoluzione di UIP (BAL-scintigrafia con Gallio 67 -IC tissutali e circolanti) v. La terapia antiinfiammatoria non si è dimostrata utile per migliorare l’evoluzione della malattia (alte dosi di corticosteroidi e di immunosoppressori in genere non sono efficaci)
Modello di normale riparazione tissutale
IPOTESI nascente: Danno epiteliale e alterato meccanismo di riparazione 1 4 2 3
IPF Clinical Presentation • IPF should be considered in all adult patients with unexplained chronic exertional dyspnea, and commonly presents with cough, bibasilar inspiratory crackles, and finger clubbing. • The incidence of the disease increases with older age, with presentation typically occurring in the sixth and seventh decades • Patients with IPF aged less than 50 years are rare; such patients may subsequently manifest overt features of an underlying connective tissue disease that was subclinical at the time IPF was diagnosed. • More men have been reported with IPF than women. • The majority of patients have a history of cigarette smoking
Natural history of IPF The majority of patients experience a slow but steady worsening of their disease (‘‘Slow progression’’). Some patients remain stable (‘‘Stable’’), while others have an accelerated decline (‘‘Rapid progression’’). A minority of patients may experience unpredictable acute worsening of their disease (lightning bolt), either from a secondary complication such as pneumonia, or for unrecognized reasons. This event may be fatal or may leave patients with substantially worsened disease. The relative frequency of each of these natural histories is unknown.
SELECTED FEATURES ASSOCIATED WITH INCREASED RISK OF MORTALITY IN IDIOPATHIC PULMONARY FIBROSIS
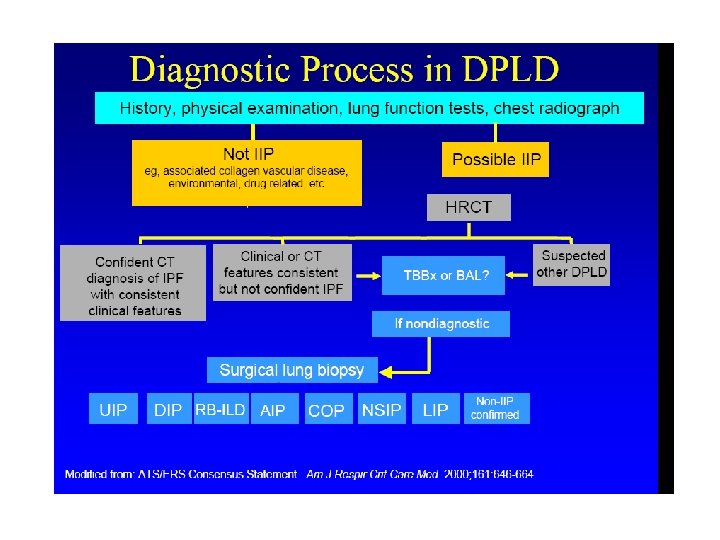
Clinical Assessment • History • Physical Exam • Pulmonary Function Testing – At Rest – Exercise • Serologic Studies • Chest Radiograph & HRCT • Tissue examination
History • • Age Gender Smoking history Medications Environmental exposure Occupational exposure Duration of symptoms Family history
Diagnostic tools in patients with suspected ILD Demographics Diseases Age >50 years Idiopathic Pulmonary Fibrosis, Cryptogenic Organizing Pneumonia (BOOP) 20 -40 years: Gender Female Male Sarcoidosis, Connective Tissue Disease Associated ILD, Lymphangioleimyomatosis, Pulmonary Langerhans Cell Histiocytosis Connective Tissue Disease Associated ILD; Lymphangioleiomyomatosis Occupational – Pneumoconiosis; Idiopathic Pulmonary Fibrosis
History: Age and Gender Age Gender (f) • • Connective Tissue Disease Associated ILD Lymphangioleiomyomatosis
History: Duration of Illness 1. Acute Diseases (Days to weeks) • DAD (AIP), EP, Vasculitis/DPH, Drug, CVD ________________________________________________________ 2. Subacute Diseases (weeks to months) • HSP, Sarcoid, Cellular NSIP, Drug, “Chronic” EP, Bronchiolitis/ SAD _________________________________________________________ 3. Chronic Diseases (months to years) • UIP, Fibrotic NSIP, Pneumoconioses,
Physical Findings • • • Resting Tachypnea Shallow breathing Dry crackles Digital clubbing Pulmonary HTN Non-pulmonary findings
Physical Findings
PFT: Restrictive Disease VC VC VC TLC RV Normal TLC RV RV ILD NM Disease
PFT Restrictive-obstructive pattern: • ILD associated with asthma or recurrent bronchospasm include: Churg-Strauss syndrome, Sarcoidosis (endobronchial) ABPA Resting pulmonary function tests: Document the existence, gauge the severity and provide clues that are useful in the differential diagnosis of ILD. are useful in the monitoring of clinical progression of the disease or response to therapy.
PFT: cardiopulmonary exercise test • Exercise affords the most sensitive diagnostic and physiological test for ILD. • The degree of arterial hypoxemia induced by exercise and the alveolar-arterial difference in P 02 (PAO 2 – Pa. O 2 gradient) correlate well with the degree of pulmonary fibrosis.
Chest X Ray: limits • CXR is normal: – in 10 to 15 % of symptomatic patients with proven infiltrative lung disease – 30% of those with bronchiectasis – ~ 60 % of patients with emphysema • CXR has a sensitivity of 80% and a specificity of 82% percent for detection of ILD • CXR can provide a confident diagnosis in ~ 23 % of cases
CXR CLUES Alveolar Filling Air-bronchograms Acinar rosettes Diffuse consolidation Nodule like, poor boarder definition • Silhouetting: obliteration of normal structures • •
CXR CLUES Interstitial Infiltrates • Nodular • Linear or reticular • Mixed • Honeycomb • Cysts and traction bronchiectasis • Ground Glass Opacities
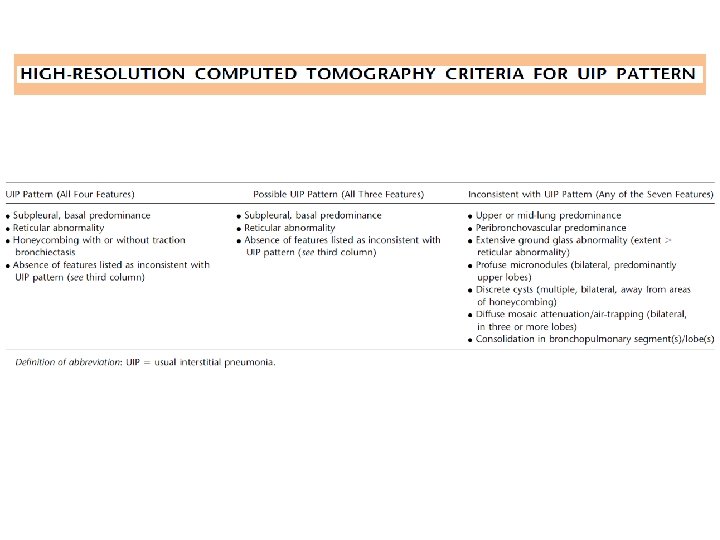
COMBINATION OF HIGH-RESOLUTION COMPUTED TOMOGRAPHY AND SURGICAL LUNG BIOPSY FOR THE DIAGNOSIS OF IPF
High-resolution computed tomography (HRCT) images demonstrating usual interstitial pneumonia (UIP) pattern and possible UIP pattern. (A and B) UIP pattern, with extensive honeycombing: axial and coronal HRCT images show basal predominant, peripheral predominant reticular abnormality with multiple layers of honeycombing (arrows). (C and D) UIP pattern, with less severe honeycombing: axial and coronal CT images how basal predominant, peripheral predominant reticular abnormality with subpleural honeycombing (arrows). (E and F ) Possible UP pattern: axial and coronal images show peripheral predominant, basal predominant reticular abnormality with a moderate amount of ground glass abnormality, but without honeycombing.
Surgical lung biopsy specimens demonstrating UIP pattern. (A) Scanning power microscopy showing a patchy process with honeycomb spaces (thick arrow), some preserved lung tissue regions (thin arrow), and fibrosis extending into the lung from the subpleural regions.
(B) Adjacent to the regions of more chronic fibrosis (thick arrow) is a fibroblast focus (asterisk), recognized by its convex shape and composition of edematous fibroblastic tissue, suggestive of recent lung injury.
BAL cellular analysis • In the evaluation of patients with suspected IPF, the most important application of BAL is in the exclusion of chronic hypersensitivity pneumonitis; prominent lymphocytosis (40%) should suggest the diagnosis. • Recommendation: BAL cellular analysis should not be performed in the diagnostic evaluation of IPF in the majority of patients, but may be appropriate in a minority • Values: This recommendation places a high value on the additional risk and cost of BAL in patients with IPF and a low value on possible improved specificity of diagnosis. • Remarks: This recommendation is only for BAL differential cell count (‘‘cellular analysis’’). It does not refer to the use of BAL in the evaluation of infection, malignancy, etc.
BAL cellular analysis
Transbronchial biopsy • While transbronchial biopsy specimens may show all the histologic features of UIP, the sensitivity and specificity of this approach for the diagnosis for UIP pattern is unknown. It is also unknown how many and from where transbronchial biopsies should be obtained. • Recommendation: Transbronchial biopsy should not be used in the evaluation of IPF in the majority of patients, but may be appropriate in a minority. • Values: This recommendation places a high value on the additional morbidity of transbronchial lung biopsy in patients with IPF who will subsequently undergo surgical lung biopsy and low value on possible diagnostic specificity.
Transbronchial biopsy Provide additional information, especially when tissue abnormalities tend to be distributed in peribronchiovascular areas e. g. Sarcoidosis, LAM and Lymphangitic carcinomatosis. It may disclose certain distinctive abnormalities e. g. Tight, uniform, well formed non caseating granulomas of sarcoidosis. Smooth muscle proliferation of LAM. Lymphatic metastasis of malignant cells. Giant cell granulomas are suggestive of hard metal pneumoconiosis. It is diagnostic if an infectious agent or malignancy is detected.
Serologic testing for connective tissues disease • There are no reliable data on the role of screening serologies in patients with suspected IPF. Connective tissue disease can present with a UIP pattern, and ILD has been described as the sole clinical manifestation of these conditions and can precede the overt manifestation of a specific connective tissue disease. • Recommendation: Serologic testing for connective tissue disease should be performed in the evaluation of IPF in the majority of patients. • Values: This recommendation places a high value on distinguishing connective tissue disease from IPF. • Remarks: Serologic evaluation should be performed even in the absence of signs or symptoms of connective tissue disease, and should include rheumatoid factor, anti-cyclic citrullinated peptide, and anti-nuclear antibody titer and pattern. The routine use of other serological tests such as antisynthetase antibodies (e. g. , Jo-1), creatine kinase and aldolase, Sjogren’s antibodies (SS-A, SS-B), and scleroderma antibodies (scl-70, PM-1) is of unclear benefit. Patients with IPF may have a mildly positive antinuclear antibody titer and/or rheumatoid factor level without any other clinical features of connective tissue disease • •
Useful laboratory tests for patients with ILD, beyond routine laboratory testing Abbreviations: ANA, antinuclear antibody; c-ANCA, cytoplasmic antineutrophil cytoplasmic antibody (antiproteinase 3); CSS, Churg-Strauss syndrome; CTD, connective tissue disease; DAH, diffuse alveolar hemorrhage; LIP, lymphocytic interstitial pneumonia; MPA, microscopic polyangiitis; p-ANCA, perinuclear antineutrophil cytoplasmic antibody (antimyeloperoxidase).
Diagnostic Criteria • 1. Exclusion of other known causes of ILD (e. g. , domestic and occupational environmental exposures, connective tissue disease, and drug toxicity). • 2. The presence of a UIP pattern on HRCT in patients not subjected to surgical lung biopsy. • 3. Specific combinations of HRCT and surgical lung biopsy pattern in patients subjected to surgical lung biopsy.
Diagnostic algorithm for IPF MDD: multidisciplinar discussion
Connective tissue diseases associated with usual interstitial pneumonitis • • Rheumatoid arthritis Systemic sclerosis (scleroderma) Polymyositis/ dermatomyositis Mixed connective tissue disease Undifferentiated connective tissue disease Systemic lupus erythematosus (rare) Primary Sjorgren’s syndrome (rare) Strange Clin Chest Med 25 (2004) 549– 559
Pulmonary diseases associated with systemic sclerosis • • • • Interstitial lung disease Fibrotic nonspecific interstitial pneumonitis Usual interstitial pneumonitis Diffuse alveolar damage (rare) Cryptogenic organizing pneumonia Pulmonary hypertension (PH) Pulmonary arterial hypertension most common in lc. SSc Secondary PH from ILD most common in dc. SSc Lung cancer Aspiration pneumonia Pleuritis and pericarditis Alveolar hemorrhage (rare) Spontaneous pneumothorax (rare) Strange Clin Chest Med 25 (2004) 549– 559
Lung diseases associated with rheumatoid arthritis • • • • Rheumatoid interstitial lung disease Non-specific interstitial pneumonitis Usual interstitial pneumonitis Cryptogenic organizing pneumonia Diffuse alveolar damage Exudative pleural effusions Rheumatoid nodules Bronchiolitis Follicular bronchiolitis Constrictive bronchiolitis Bronchiolitis obliterans Bronchiectasis Methotrexate pneumonitis Gold pneumonitis Cricoarytenoid arthritis with upper airway obstruction Strange Clin Chest Med 25 (2004) 549– 559
Most frequent pulmonary complications of systemic lupus erythematosus Lupus pleurisy Lupus pneumonitis Shrinking lung syndrome Bacterial pneumonia Immune modulating drugs Altered immunity Capillaritis with alveolar hemorrhage Pulmonary emboli associated with lupus anticoagulants • Pulmonary arterial hypertension • •
Symptoms associated with connective tissue diseases Joint and muscle symptoms • Arthritis • Arthralgias • Morning stiffness • Synovitis • Myalgias • Muscle weakness • Neuropathy Raynaud’s syndrome Gastrointestinal symptoms • Esophageal reflux • Dysphagia • Abdominal bloating, diarrhea Sicca symptoms • Dry eyes • Dry mouth Strange Clin Chest Med 25 (2004) 549– 559 Skin manifestations • Malar rash • Photosensitivity • Machinist’s hands • Gottron’s papules • Erythema nodosum • Subcutaneous nodules • Sclerodactyly • Digital or oral ulcers • Edema • Telangiectasias Chest pain • Pleurisy • Pericarditis Eye abnormalities • Corneal ulcers • Uveitis • Scleritis