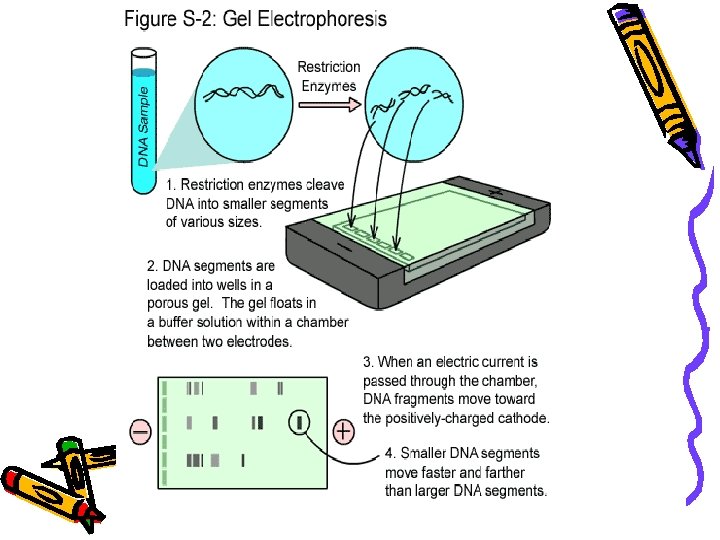
Gel Electrophoresis Agarose Gel Electrophoresis Electro flow of











. The agarose solution is boiled")
 • The buffer provides ions")
















 Anode (negative) Add enough electrophoresis buffer to cover the")





 wells Bromophenol Blue DNA (-) Gel Anode (+) After the current is")




 DNA ladder 1 2 3 4 5 6 7")



















 •")












- Slides: 74
Gel Electrophoresis
Agarose Gel Electrophoresis • Electro = flow of electricity, phoresis, from the Greek = to carry across • is an analytical technique used to separate DNA, RNA and Protein fragments by size. • An electric field forces the fragments to migrate through a gel.
• DNA molecules normally migrate from negative to positive potential due to the net negative charge of the phosphate backbone of the DNA chain. • Longer molecules migrate more slowly because they are more easily 'trapped' in the network.
• DNA is negatively charged. • When placed in an electrical field, DNA will migrate toward the positive • An agarose gel is used to slow the movement of DNA and pole (anode). separate by size. H O 2 DNA - Power + • Polymerized agarose is porous, allowing for the movement of DNA Scanning Electron Micrograph of Agarose Gel (1× 1 µm)
How fast will the DNA migrate? …gel electrophoresis separates DNA according to size DNA - Power small large + Within an agarose gel, linear DNA migrate inversely proportional to the log 10 of their molecular weight.
Gel Electrophoresis
What is needed? • The types of gel most commonly used for DNA electrophoresis are Agarose
Agarose D-galactose 3, 6 -anhydro L-galactose • Agarose was first used in biology when Robert Koch* used it as a culture medium for Tuberculosis bacteria in 1882 Agarose is a linear polymer polysaccharide extracted from seaweed.
• Agarose (for relatively long DNA molecules 200 to 50, 000 base pairs long). • Agarose is dissolved in buffer and heated, then cools to a gelatinous solid with a network of crosslinked molecules start end
An agarose gel is prepared by combining agarose powder and a buffer solution. Buffer polyacrylamide (for high resolution of short DNA molecules (Plasmid), for example in DNA sequencing) When sharper bands are required. Flask for boiling Agarose
Agarose Buffer Solution Combine the agarose powder and buffer solution. Use a flask that is several times larger than the volume of buffer.
Melting the Agarose is insoluble at room temperature (left). The agarose solution is boiled until clear (right). *Be careful when boiling - the agarose solution may become superheated and may boil violently if it has been heated too long in a microwave oven.
• Buffer - in this case TAE (TBE) • The buffer provides ions in solution to ensure electrical conductivity. Tris-acetate buffer (TAE) usually at p. H 8. 0, and EDTA, which sequesters divalent cations. TAE has a lower buffer capacity than TBE and easily can become exhausted, but linear, double stranded DNA runs faster in TAE
• Also needed are a power supply • Gel chambers come in a variety of models, from commercial through homemade, and a variety of sizes
How does it work? • DNA is an organic acid, and is negatively charged (remember, DNA for Negative) • When the DNA is exposed to an electrical field, the particles migrate toward the positive electrode • Smaller pieces of DNA can travel further in a given time than larger pieces
DNA negatively charged
Steps in running a gel • DNA is prepared by digestion with restriction enzymes • Agarose is made to an appropriate thickness (the higher the % agarose, the slower the big fragments run) and ‘melted’ in the microwave • The gel chamber is set up, the ‘comb’ is inserted • The agarose may have a DNA ‘dye’ added (or it may be stained later). The agarose is poured onto the gel block and cooled
DNA cut up using restriction endonucleases
Electrophoresis Equipment Power supply Gel tank Cover Electrical leads Casting tray Gel combs
Gel casting tray & combs
Make up the gel which the DNA will be put into
• The comb is removed, leaving little ‘wells’ and buffer is poured over the gel to cover it completely • The DNA samples are mixed with a dense loading dye so they sink into their wells and can be seen
Dye added to the DNA Bromophenol Blue + Sucrose
Buffer solution added to the tank
DNA samples loaded into wells
• The DNA samples are put in the wells with a micropipette. • Micropipettes have disposable tips and can accurately measure 1/1, 000 of a liter
DNA buffer wells Cathode (positive) Anode (negative) Add enough electrophoresis buffer to cover the gel to a depth of at least 1 mm. Make sure each well is filled with buffer.
Running the Gel Place the cover on the electrophoresis chamber, connecting the electrical leads. Connect the electrical leads to the power supply. Be sure the leads are attached correctly - DNA migrates toward the cathode (red). When the power is turned on, bubbles should form on the electrodes in the electrophoresis chamber.
A gel being run Positive electrode Comb Agarose block DNA loaded in wells in the agarose Black background To make loading wells easier Buffer
Loading the Gel Carefully place the pipette tip over a well and gently expel the sample. The sample should sink into the well. Be careful not to puncture the gel with the pipette tip. (5µlof sample +3µlof dye)
Place the gel in the electrophoresis chamber.
Next? • The power source is turned on and the gel is run. • The time of the run depends upon the amount of current and % gel, and requires experimentation • At the end of the run the gel is removed (it is actually quite stiff) • The gel is then visualized - UV light causes the bands of DNA to fluoresce
Cathode (-) wells Bromophenol Blue DNA (-) Gel Anode (+) After the current is applied, make sure the Gel is running in the correct direction. Bromophenol blue will run in the same direction as the DNA.
DNA Ladder Standard 12, 000 bp 5, 000 DNA migration bromophenol blue 2, 000 1, 650 1, 000 850 650 500 400 300 200 100 + Inclusion of a DNA ladder (DNAs of know sizes) on the gel makes it easy to determine the sizes of unknown DNAs.
Staining the Gel • Ethidium bromide binds to DNA and fluoresces under UV light, allowing the visualization of DNA on a Gel. • Ethidium bromide can be added to the gel and/or running buffer before the gel is run or the gel can be stained after it has run. ***CAUTION! Ethidium bromide is a powerful mutagen and is moderately toxic. Gloves should be worn at all times.
Staining the Gel • Place the gel in the staining tray containing warm diluted stain. • Allow the gel to stain for 25 -30 minutes. • To remove excess stain, allow the gel to destain in water. • Replace water several times for efficient destain.
Ethidium Bromide requires an ultraviolet light source to visualize
Visualizing the DNA (ethidium bromide) DNA ladder 1 2 3 4 5 6 7 8 wells 5, 000 bp 2, 000 1, 650 1, 000 850 650 500 400 300 200 100 PCR Product Primer dimers + - - + + Samples # 1, 4, 6 & 7 were positive for DNA
wells DNA ladder PCR Product + - - + 2, 000 bp 1, 500 1, 000 750 500 250 Samples # 1, 6, 7, 10 & 12 were positive for DNA March 12, 2006
A gel as seen under UV light - some samples had 2 fragments of DNA, while others had none or one
DNA is stained using ethidium bromide
More…… • Many samples can be run on one gel- but it is important to keep track • Most gels have one lane as a ‘DNA ladder’ - DNA fragments of known size are used for comparison
• Based this info, how big is the circled fragment? – 850 bp 1000 bp 850 bp 750 bp 600 bp 200 bp 100 bp + St an Sa mp dard l Sa e 1 mp le 2 - Si • Suppose you have a size standard with the following sized fragments: 1000 bp, 850 bp, 750 bp, 600 bp, 200 bp, 100 bp ze Example
Still more…. • The DNA band of interest can be cut of the gel and the DNA extracted - • Or DNA can be removed from the gel by Southern Blotting
References www. biotech. iastate. edu/publication/pptpresentations Kreuzer, H. , Massey, A. , 2001, Recombinant DNA and Biotechnology, 2 nd ed. ASM Press, Washington Turner, P. C. , et al, 1997, Instant Notes in Molecular Biology, Bios, Oxford Photos - L. D. Macdonald, 2003
This powerpoint was kindly donated to www. worldofteaching. com http: //www. worldofteaching. com is home to over a thousand powerpoints submitted by teachers. This is a completely free site and requires no registration. Please visit and I hope it will help in your teaching.
• Prepare a Stock Solution of EDTA • • • 0. 5 M Na 2 EDTA (ethylenediamine tetraacetic acid) PH 8. 0 18. 6 gr EDTA 80 ml deionized water Adjust the PH with Na. OH D/water up to 100 ml
• Prepare a Stock Solution of TAE • • • For 50 ml 0 f 50 X TAE buffer: 14. 1 gr Tris Base (2 -amino-2 -hydroxymethyl-propane-1, 3 -diol) 18. 5 ml glacial acetic acid 5 ml EDTA D/W up to 50 ml • Prepare a Working Solution of TAE • For agarose gel electrophoresis, TAE can be used at a concentraion of 1 x (1: 50 dilution of the concentrated stock). • Dilute the stock solution deionized water • Finally 1 ml TAE add 49 ml D/W • Be stored at room temperature
• Prepare a Stock Solution of TAE • for lab work for each group • For 5 ml 0 f 50 X TAE buffer: • 1. 41 gr Tris Base (2 -amino-2 -hydroxymethyl-propane-1, 3 -diol) • 1. 85 ml glacial acetic acid • 0. 5 ml EDTA (0. 186 gr EDTA + 0. 8 ml D/Water adjust Ph to 8 by Na. OH add D/water up to 1 ml) • D/W up to 5 ml
Prepare Agarose Gelland Casting Tray • 1 -For a 2% agarose gel, weigh out 2 g of agarose into a flask and add 100 ml of 1 x TBE or TAE. • 2 -Heat solution in a microwave or boiling water bath until agarose is completely dissolved. • 3 -Allow to cool in a water bath set at 50 – 55°C for 10 min. • 4 -Prepare gel casting tray by sealing ends of gel chamber with tape or appropriate casting system. Place appropriate number of combs in gel tray.
Prepare of working solutionof TAE For agarose gel electrophoresis, TAE can be used at a concentration of 1 x (1: 50 dilution of the concentrated stock). Dilute the stock solution deionized water Finally 1 ml TAE add 49 ml D/W Be stored at room temperature
Prepare Agarose Gelland Casting Tray • 5 -cooled gel and pour into gel tray. Allow to cool for 15 -30 min at room temperature. Gels can also be placed in a cold space and used the following day. • 6 -Remove comb(s), place in electrophoresis chamber and cover with buffer (TAE or TBE as used previously). • 7 - ((Add 5 ul of ethidium bromide 0. 5 μg/ml working solution))
Loading Dye • Adds loading dye (Bromophenol Blue 25 mg, + to the DNA sample so that it will go into the 10 -20 ul well (1 ul dye 1 X : 5 ul DNA) Sucrose 4 g and. H 2 O up to 10 ml) – makes it sink to the bottom • It Adds blue color so you can see what you are pipetting
Loading DNA • 1 -Add loading buffer to samples. As a guideline, add 1. 5 ul of 10 x Loading Buffer to a 20 -25 ul PCR/DNA solution. For more concentrated DNA solutions (e. g. plasmids), prepare tubes with 8 ul of 1 x Loading Buffer and 2 ul of DNA. • 2 -Add 1: 5, Loading dye: DNA • 3 -Load DNA and standard (Ladder) onto gel. • 4 -Electrophorese at 100 V for 1 h. • 5 -Visualize DNA bands using UV lightbox or gel imaging system.
Purpose of Gel Electrophoresis • A method for separating DNA • Can be used to separate the size of – DNA – RNA – Protein • We will be using it to separate DNA
DNA • What you start with: A variety of different fragments of DNA all mixed together • We will use gel electrophoresis to separate/sort these fragments
How It Separates • The gel is a porous matrix (like a sponge) • Separates DNA based on – Size – Charge
Separation by Size • As DNA is moved through the gel, smaller sized fragments move through faster than larger sized fragments – Ex. A 100 base pair fragment will move through the gel faster than a 500 bp fragment start end Image taken without permission from http: //www. dnai. org/b/index. html-- Gel Electrophoresis Animation
Separation Using Charge • The charge on DNA is what makes it move through the gel • DNA is a charged molecule. What is the charge on DNA? – Negative charge • Why? – Phosphate group is negatively charged Image taken without permission from http: //www. dnai. org/b/index. html-- Gel Electrophoresis Animation
Separation Using Charge • The gel is hooked up to a power source • DNA is loaded into the gel on the cathode (-) end • Gel is placed in a buffer solution that will conduct electricity • Electric current is run through the gel – DNA is attracted to the + end (anode) – “runs to the Image taken without permission from http: //www. dnai. org/b/index. html-- Gel Electrophoresis Animation
The Gel • Wells are created to put the DNA into • We use agarose gels to separate DNA wells - - well Direction DNA travels + Direction DNA travels SIDE VIEW TOP VIEW +
Challenges • DNA is colorless-- how will we see it on the gel & when we are loading it into the gel? • How do we get the DNA to stay in the well (not float away)?
Solution #1 • Problem #1: How can we see the DNA sample as we load it into the gel • Problem #2: How can we make sure DNA won’t float away • Solution: Add loading dye to the initial DNA sample!
Loading Dye • Adds mass to the DNA sample so that it will go into the well – makes it sink to the bottom • Adds blue color so you can see what you are pipetting
Solution #2 • Problem: DNA is colorless. Once the DNA has been run through the gel, how can we see where it is on the gel? • Solution: Add Ethidium Bromide (Et. Br) to the gel
Ethidium Bromide • The DNA intercalates with the Ethidium Bromide – Intercalates = inserts itself between bases • Et. Br will fluoresce under UV light
Relative Size vs. Absolute Size • Looking at a gel, you can determine which fragments of DNA are bigger than others = Relative Size • Which fragment is bigger, A or B? – Fragment A (didn’t travel as far in a fixed amount of time) (-) start A B (+) end
Absolute Size • How can we determine the actual size of the DNA fragments (how many base pairs- bp)? • Use a size standard – Also called a DNA ladder – Consists of a series of fragments of known sizes – Use it to compare to your DNA fragments
• Based this info, how big is the circled fragment? – 850 bp 1000 bp 850 bp 750 bp 600 bp 200 bp 100 bp + St an Sa mp dard l Sa e 1 mp le 2 - Si • Suppose you have a size standard with the following sized fragments: 1000 bp, 850 bp, 750 bp, 600 bp, 200 bp, 100 bp ze Example