ELECTROPHORESIS Daheeya Al Enazi CLS 332 PRINCIPLES OF



")
 A charged particle (+Q) moving in an electric field (E) in a")












 : is a method of")

 for")




,")


, PH=8. 8")





- Slides: 35
ELECTROPHORESIS Daheeya Al. Enazi CLS 332
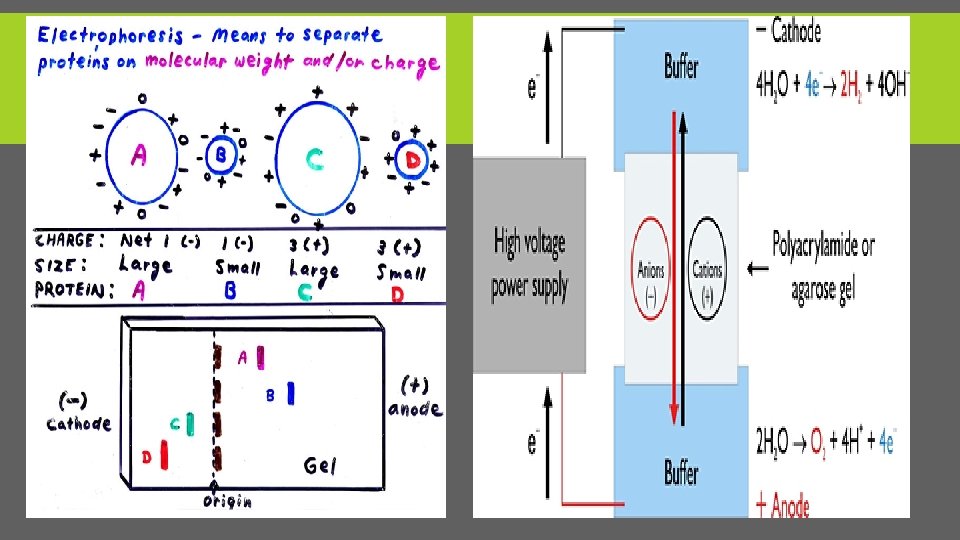
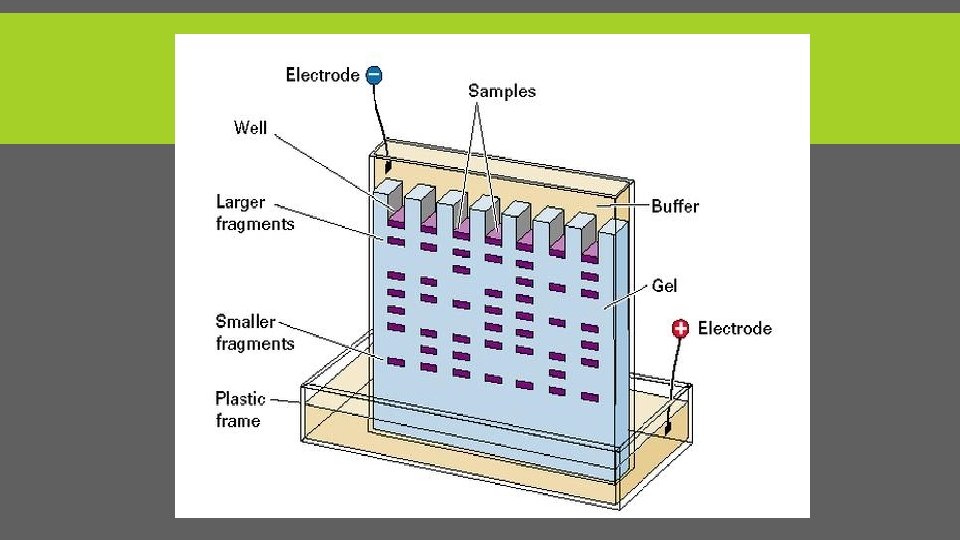
PRINCIPLES OF ELECTROPHORESIS § Electrophoresis is a method used to separate charged particles from one another based on differences in their migration speed according in to there charge and size(m. wt). § This is achieved by moving negatively charged molecules (protein and nucleic acid) through a matrix (support medium )with an electric field. § Smaller molecules move faster and migrate farther than bigger ones.
SUPPORT MEDIUM 1. a strip of paper or cellulose acetate (i. e. when small peptides are to be separated). 2. Gel, such as Agarose and polyacrylamide (commonly used)
ELECTROPHORESIS PARTS There are two electrodes (made of an inert metal, e. g. platinum) are immersed in two separate buffer chambers. By using an electric power supply, electric potential (E) is generated between the two electrodes. Charged particles migrate from one chamber to the other according to it charge. Negatively charged ions, called anions, move towards the positively charged anode, while positively charged ions, called cations, move towards the negative charged cathode. Different ions migrate at different speeds by their sizes and by the number of charges they carry. As a result, different ions can be separated from each other by electrophoresis
FORCES (F) A charged particle (+Q) moving in an electric field (E) in a non conducting medium, such as water. Two forces are exerted on the particle, one Fe, the force exerted on the charged particle by the field, which is in the direction of the motion (toward the cathode), and the other, Ff, the frictional force on the charged particle, which retards its motion toward the cathode, and hence is in the direction opposite to the motion (toward the anode (+) electrode). This is shown in the diagram below:
CRITERIA OF GEL: 1. Hydrophilic 2. Chemically stable (not participate in chemical reactions during electrophoresis) 3. Neutral (free of electric charges, otherwise it would act as an ion exchanger) 4. Mechanically resistant (should not be too elastic or too rigid as such gels would be difficult to handle). 5. Bands need to be visualized in the gel by using special stain, the gel should be transparent, 6. Pore size of the gel can be controlled during the preparation of the gel.
EFFECT OF PH ON MOBILITY 1. The effective charge of a substance varies with the p. H also its effective mobility varies. 2. Electrophoretic solutions must be buffered to carry the electric field. 3. The buffer p. H is chosen to give an optimum net charge for maximum separation.
*AGAROSE GEL ELECTROPHORESIS Mainly used in analysis or separation of DNA , RNA molecules and protein. Molecules move according their size & net charge. v. Criteria: ØAdvantages: Gel is easily poured, does not denature the samples and samples can also be recovered. ØDisadvantages : gels can melt during electrophoresis, the buffer can become exhausted, and different forms of genetic material may run in unpredictable forms.
PREPARATION OF GEL A-Percentage of agarose %: -Between 0. 7% - 2%. 0. 7% good separation or resolution of large 5 – 10 kb DNA fragments. 2% good resolution for small 0. 2– 1 kb fragments. Agarose dissolved in electrophoresis buffer. B-Buffer: Tris/Borate/EDTA (TBE) DNA Tris/Acetic acid/EDTA protein
ANALYSIS ØAfter electrophoresis the gel is illuminated with an ultraviolet lamp to view the DNA bands. ØThe ethidium bromide fluoresces reddish-orange in the presence of DNA. (Carcinogenic stain ) Øphotograph it with a digital camera. Our lab: üUse program analysis to analyze the gel and take picture of gel.
LAB PREPARATION 1. Make a 2% agarose solution in 100 ml TAE 2 g of agarose+100 ml of TAE buffer. 2. put in microwave for 1 - 1 ½ min. to dissolve the agarose. 3. Let it cool down to about 55 - 60 C at RT. 4. Add ethidium bromide stock in the gel solution for a final concentration of 0. 5 ug/ml. Be very careful when handling the concentrated stock. 5. Stir the solution to disperse the ethidium bromide, then pour it into the gel rack. 6. Insert the comb at one side of the gel, about 5 -10 mm from the end of the gel. 7. Put gel into a tank with TAE. Gel must be completely covered with TAE, with the slots at the end electrode that will have the negative current.
CONT. After the gel has been prepared, use a micropipette to inject about 3µl of stained DNA (a DNA ladder is also highly recommended). Close the lid of the electrophoresis chamber and apply current (typically 100 V for 30 minutes). The colored dye in the DNA ladder (molecular weight markers) and DNA samples acts as a "front wave" that runs faster than the DNA itself. When the "front wave" approaches the end of the gel, the current is stopped. The DNA is stained with Et. Br, and is then visible under ultraviolet light.
APPLICATION OF AGAROSE GEL 1. Separation Of DNA 2. Analysis of PCR products: e. g. in molecular genetic diagnosis or genetic fingerprinting
PAGE Daheeya Al. Enazi
POLYACRYLAMIDE GEL ELECTROPHORESIS ØTechnique widely used in biochemistry, genetics & molecular biology to separate biological macromolecules (proteins or nucleic acids) according to their electrophoretic mobility. ØElectrophoretic mobility is a function of the length, conformation and charge of the molecule. ØPolyacrylamide gel with small pores helps to examine smaller molecules better since the small molecules can enter the pores and travel through the gel while large molecules get trapped at the pore openings.
ACRYLAMIDE ØAcrylamide monomer is in a powder state before addition of water. ØAcrylamide is toxic to the human nervous system, therefore all safety measures must be followed when working with it. ØAcrylamide is soluble in water and upon addition of water it polymerizes resulting in formation of polyacrylamide.
A-NATIVE-PAGE Gel electrophoresis, molecules may be run in their native state, preserving the molecules' higher-order structure. Protein structure
B-SDS PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) : is a method of separating molecules based on the difference of their molecular weight. SDS chemical denaturant or detergent can destroys the protein secondary, tertiary or quaternary structure and turning them into negatively charged linear polypeptide chains. At certain p. H the SDS molecules are negatively charged and bind to proteins in a set ratio, approximately one molecule of SDS for every 2 amino acids. SDS is applies a negative charge to each protein in proportion to its mass. After running the electric field , the negatively charged polypeptide chains travel toward the anode with different mobility.
The result: The mobility of the molecules, is inversely proportional to the logarithm of their molecular weight. Use Tracking Dye (SDS dye): To determined the distance traveled by molecule.
TYPE OF DENATURANT 1. Urea For nucleic acids. 2. Sodium dodecyl sulfate (SDS) for protein. Ø 2 -Mercaptoethanol(2 Mp. E): Reducing agent used to disrupt the disulfide bonds found between the protein complexes, which helps further denature the protein.
GOOD PAGE Using both Native and SDS-PAGE together can be used to purify and to separate the various subunits of the protein. WHY? ? ØNative-PAGE keeps the oligomeric form(quaternary protein structure) intact and will show a band on the gel that is representative of the level of activity. ØSDS-PAGE will denature and separate the oligomeric form into its monomers, showing bands that are representative of their molecular weights. ØThese bands can be used to identify and assess the purity of the protein
Procedure
A- SAMPLE PREP. *Prepare loading buffer by Adding: 1. SDS to break down the protein structure 2. Glycerol to give viscous medium to sample 3. Bromophenol blue Tracking dye (Sample dye gives blue color ) 4. 2 -Mp. E to break down the disulfide bonds of protein. *Add 15 ul of loading buffer + 15 ul of protein sample. *Mix in vortex, put in hot plate for 5 min at 95 C then centrifuge for 1 min *then, samples are ready to load in to gel.
SAMPLE PREP. 2 -mercaptoethanol
B-PREPARING ACRYLAMIDE GELS Gels consist of : 1. acrylamide, bis-acrylamide, denaturant (SDS or urea), and buffer with an adjusted PH. (6. 8 -8. 8) 2. Then, solution may be degassed under a vacuum or add butanol (in case of protein)to prevent the formation of air bubbles during polymerization and get smooth surface. 3. Ammonium persulfate and TEMED as source of free radicals and a stabilizer and to initiate polymerization.
POLYMERIZATION REACTION ØReact 1 acrylamide with 35 bis-acrylamide, which can form cross-links between two acrylamide molecules. ØAcrylamide concentration from 5% - 25%. ØLower % better for resolving very high M. wt. , while higher % of acrylamide are needed to resolve smaller proteins.
POLYMERIZATION STEPS 8. 8 for separation gel 6. 8 for stacking gel
Add separation and stacking gel ØFirst layer: Separation gel (7 -15% acrylamide), PH=8. 8 good for separation the molecules. ØSecond layer: Isopropanol or ethanol to make flat surface. Put for sec. then, pour out. ØThird layer: Stacking gel(2 -4% acrylamide), PH=6. 8 use this PH because the N-terminal amino group of the proteins and amino acids are protonated at equilibrium which makes them less negative. The average electrophoretic mobility is very slow. The result is line up all samples in same level. ØAfter polymerizing put the gel between two glass plates in a gel caster, with a comb inserted at the top to create the sample wells. After the gel is polymerized the comb can be removed and the gel is ready for electrophoresis
DISTRIBUTION OF GELS
START ELECTROPHORESIS 1. Put SDS dye. In 1 st well 2. Load the samples. 3. Put the running buffer. 4. Start the electrical field for 5 min. & 50 Volt to line up all the samples at same level. (Function of Stacking gel) 5. then, adjust the machine into 40 min & 150 Volt to start separation. 6. Fixation the gel( methanol + acetic acid) for 15 min. 7. Stain with Colloidal Coomassie stain for 1 hr. 8. De-stain with 100 DW for 15 min then change the water. Repeat for 4 times or keep for overnight. 9. Take pic. by software.
The gel: Either ventricle for protein or horizontal for DNA and RNA.
FINAL RESULT Bromophenol blue SDS Dye