NEUROPATIE OBWODOWE Hanna Drac Klinika Neurologii Akademii Medycznej

NEUROPATIE OBWODOWE Hanna Drac Klinika Neurologii Akademii Medycznej w Warszawie

NEUROPATIE OBWODOWE • 3. 4 -3. 6 % populacji w starszym wieku • 50 % chorych w oddziałach ogólnych • 10 -20 % etiologia nieznana – zwykle są to aksonopatie o łagodnym przebiegu.

NEUROPATIE AKSONALNE 20 % ETIOLOGIA NIEZNANA PRZEWLEKŁE NEUROPATIE AKSONALNE NABYTE TOKSYCZNE GENETYCZNIE UWARUNKOWANE METABOLICZNE W CHOROBACH UKŁADOWYCH
 NIEDOBOROWE (B 1, B")
PRZEWLEKŁE NEUROPATIE AKSONALNE • • METABOLICZNE (cukrzyca, mocznica, nadczynność tarczycy) NIEDOBOROWE (B 1, B 12, E) TOKSYCZNE (przemysłowe, leki) CHOROBY TKANKI ŁĄCZNEJ (RA, z. Sjögrena, krioglobulinemia) INNE UKŁADOWE (paranowotworowe, sarkoidoza) ZAKAŹNE (borelioza, HIV) PARAPROTEINEMIE (MGUS, szpiczak) DZIEDZICZNE (CMT 2, amyloidoza)

MECHANIZMY NEUROPATII • • Zaburzenia metaboliczne Mechanizmy immunologiczne Niedokrwienie nerwu Zaburzenia genetyczne

OBJAWY NEUROPATII + + + - -
 ü")
NEUROPATIE BÓLOWE – NEUROPATIE MAŁYCH WŁÓKIEN śr<7µm OBJAWY: ü BÓL SPONTANICZNY (parestezje, dyzestezje) ü BÓL WYWOŁANY (allodynia) ü HYPERALGEZJA NABYTE NEUROPATIE ü ü ü ü cukrzyca paraproteinemie toksyczne niedoborowe (alc) zespół G-B CIDP HIV, borelioza zespół Sjögrena GENETYCZNE NEUROPATIE ü ü ü HSAN amyloidoza choroba Fabry’ego

KLINICZNE KATEGORIE NEUROPATII • SYMETRYCZNE ZAJĘCIE NERWÓW polineuropatia, poliradikuloneuropatia. • USZKODZENIE POJEDYŃCZEGO NERWU mononeuropatia • USZKODZENIE KILKU NERWÓW mononeuropatia mnoga mononeuropatia wieloogniskowa

PRZYCZYNY MONONEUROPATII MNOGIEJ ü Cukrzyca ü Pierwotne i wtórne układowe vasculitis ü Chłoniaki ü Borelioza ü Sarkoidoza ü Neuropatia ruchowa z blokiem przewodzenia ü Wieloogniskowa neuropatia czuciowo-ruchowa ü Krioglobulinemia ü Dziedziczna neuropatia z ucisku ü Nieukładowe vasculitis w nerwie
 PRZEBIEG")
ROZPOZNANIE NEUROPATII • • • DOKŁADNY WYWIAD!! POCZĄTEK OBJAWÓW (ostry? podostry? przewlekły? ) PRZEBIEG NEUROPATII (szybko – powoli postępujący? , zwalniająco – nawracający? KATEGORIA OBJAWÓW SUBIEKTYWNYCH ruchowe? czuciowe? mieszane? OBECNE I PRZEBYTE CHOROBY, UŻYWKI (LEKI) WYWIAD RODZINNY

NEUROPATIE O OSTRYM POCZĄTKU • • zespół G-B porfiria neuropatia stanu krytycznego idiopatyczna neuropatia czuciowa

DIAGNOSTYKA NEUROPATII – OCENA KLINICZNA • • ROZKŁAD NIEDOWŁADU I ZANIKU MIĘŚNI ROZKŁAD UBYTKU ODRUCHÓW ROZKŁAD ZABURZEŃ CZUCIA RODZAJ ZABURZEŃ CZUCIA Zajęcie małych włókien? Zajęcie dużych włókien? Zajęcie obu rodzajów włókien? WADY KOSTNE (stopy, kręgosłupy, podniebienie)

NEUROPATIA - BADANIA DODATKOWE ELEKTROFIZJOLOGICZNE • Badanie szybkości przewodzenia we włóknach czuciowych i ruchowych • Zapis z mięśni dosiebnych i odsiebnych kończyn • Badanie wywołanych potencjałów czuciowych (somatosensorycznych) • Badanie wywołanych potencjałów skórnych • Analiza zmienności interwału R-R

DIAGNOSTYKA ELEKTROFIZJOLOGICZNA NEUROPATII • • Topografia uszkodzenia Neuropatie czysto czuciowe ! Neuropatie czysto ruchowe ! (różnicowanie morfologiczne) neuropatie demielinizacyjne (8%), neuropatie aksonalne JEŚLI PRZYCZYNA PRZEWLEKŁEJ NEUROPATII JEST ZNANA BADANIE ELEKTROFIZJOLOGICZNE NIE JEST KONIECZNE.
")
NEUROPATIE – BADANIE PŁYNU M-RDZENIOWEGO • • • neuropatie demielinizacyjne neuropatie infekcyjne (borelioza, HIV) chłoniak W PRZEWLEKŁYCH AKSONALNYCH NEUROPATIACH NL JESTDIAGNOSTYCZNIE NIEPRZYDATNE

NEUROPATIE – BADANIA DODATKOWE PRZYCZYNA NEUROPATII OCZYWISTA: cukrzyca, mocznica, alkoholizm, leki PRZEBIEG: typowy NIE WYKONYWAĆ DALSZYCH BADAŃ DODATKOWYCH

NEUROPATIE PRZEWLEKŁE BADANIA „PIERWSZEJ LINII” pełna morfologia , OB, wit. B 12, kwas foliowy, glukoza, parametry nerkowe, parametry wątrobowe, TSH BADANIA „DRUGIEGO ETAPU” proteinogram, immunoelektroforeza, ACE, czynnik Rh, ANA, ANCA, anty – Ro, anty –La, anty HCV, HBS, anty – Hu, (porfiryny), rtg klatki piersiowej. BADANIA „ TRZECIEGO ETAPU” – ZALEŻNE OD ENG I EMG białko monoklonalne, anty – MAG, anty – GM 1, badanie DNA, biopsja nerwu.

BIOPSJA NERWU – WSKAZANA • ZAPALENIA NACZYŃ – układowe – nieukładowe • PROCESY SPICHRZENIOWE • AMYLOIDOZA • (CELE BADAWCZE)

POWIKŁANIA ZWIĄZANE Z BIOPSJĄ NERWU • • infekcja rany miejscowe zapalenie żył bolesne przewlekłe parestezje nerwiaki
 GENETYCZNIE UWARUNKOWANE (CMT, HNPP)")
POLINEUROPATIE STANOWIĄCE SAMODZIELNY ZESPÓŁ CHOROBOWY I. II. NABYTE (G-B, CIDP) GENETYCZNIE UWARUNKOWANE (CMT, HNPP)

POLINEUROPATIE W CHOROBACH OGÓLNOUSTROJOWYCH I. W CHOROBACH METABOLICZNYCH II. W CHOROBACH ZAKAŹNYCH III. W NOWOTWORACH ZŁOŚLIWYCH IV. W CHOROBACH KRWI V. W ZATRUCIACH VI. W KOLAGENOZACH VII. W PIERWOTNYCH ZAPALENIACH NACZYŃ

, Tooth (1886) Dziedziczna neuropatia ruchowo-czuciowa (Hereditary Motor")
Neuralny strzałkowy zanik mięśni Charcot, Marie (1886), Tooth (1886) Dziedziczna neuropatia ruchowo-czuciowa (Hereditary Motor Sensory Neuropathy. HMSN), Thomas i wsp. 1974; Dyck 1975 Choroba Charcot-Marie-Tooth – CMT (genetycy – lata 80 -te XX w. ) Występowanie: 1: 2500 osób

CMT podział elektrofizjologiczno-morfologiczny CMT postacie demielinizacyjne postacie pośrednie postacie aksonalne
 hipomielinizacyjne (congenital hypomyelinating neuropathy, CHN) dysmielinizacyjne (pofałdowana mielina")
Postacie demielinizacyjne przerostowa (hypertrophic neuropathy HN) hipomielinizacyjne (congenital hypomyelinating neuropathy, CHN) dysmielinizacyjne (pofałdowana mielina FFN, tomakule)
 genetycznie CMT AD PMP 22, MPZ, LITAF, EGR")
CMT Neuropatia heterogenna: klinicznie (elektrofizjologicznie, morfologicznie) genetycznie CMT AD PMP 22, MPZ, LITAF, EGR 2, KIF 1 b, MFN 2, GARS, NEFL, HSPB 1, 12 q 23 -24, 3 q 31 RAB 7, CMT AR GDAP 1, MTMR 2, SBF 2, KIAA, NDRG 1, EGR 2, PRX, MPZ, PMP 22, 10 q 23 Russe, 8 q 21. 3 CMTX (AD, AR) CX 32 DICMT 10 q 24. 1 Poznano (2005) 30 genów związanych przyczynowo z chorobą

CMT Różne mutacje w tym samym genie mogą powodować różny fenotyp choroby Przykłady MPZ, PMP 22, GDAP 1, LMNA A/C

mutacje w genie koneksyny


")
DZIEDZICZNE NEUROPATIE CZUCIOWE I AUTONOMICZNE (HSAN)


LECZENIE PORFIRII zapobieganie atakom niepodawanie większości leków – mogą wywołać napad porfirii postępowanie w napadzie hematyna (normosan) glukoza (200 -400 g/dobę) propranolol dostarczanie białka (respirator)














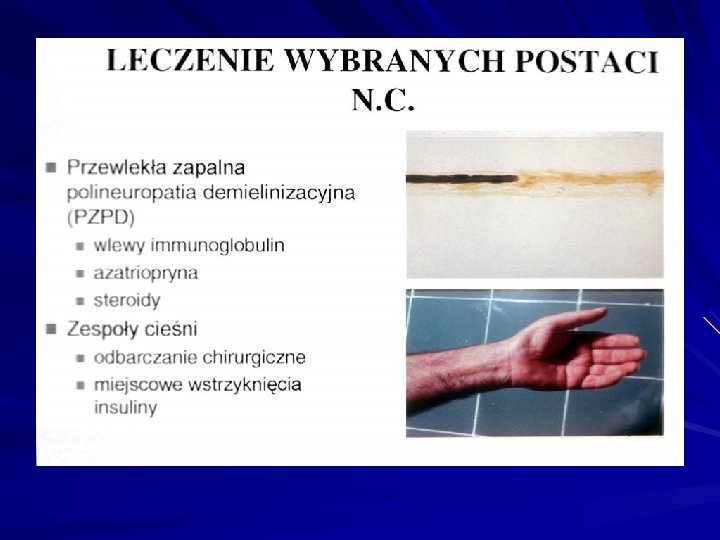
 - schemat leczenia Encorton 1 mg/kg Azathiopryna 1")
Przewlekła zapalna polineuropatia demielinizacyjna PZPD (CIDP) - schemat leczenia Encorton 1 mg/kg Azathiopryna 1 mg/kg Endoksan Immunoglobuliny Plazmafereza Rehabilitacja

Leczenie powtarzane wlewy immunoglobulin

 • Z przewagą objawów czuciowych • Polineuropatia z ubytkami")
ODSIEBNA, SYMETRYCZNA POLINEUROPATIA CUKRZYCOWA (OSPC) • Z przewagą objawów czuciowych • Polineuropatia z ubytkami lub opacznymi zaburzeniami czucia skórnego, i miernego stopnia zab. autonomicznymi oraz ruchowymi (najczęstsza postać) • Ostra, bólowa neuropatia (‘acute, painful neuropathy) • Postać ataktyczna (‘pseudotabes diabetica’) • Z przewagą zaburzeń autonomicznych • Mieszana: czuciowo-autonomiczno-ruchowa • Przewlekła zapalna polineuropatia demielinizacyjna (PZPD)








PATOLOGIE POZAWĄTROBOWE W ZAKAŻENIU WIRUSEM ZAPALENIA WĄTROBY TYPU C • • • krioglobulinemia zaburzenia funkcji tarczycy autoimmunologiczne zapalenie wątroby zespoły dermatologiczne uszkodzenie nerek zaburzenia neurologiczne – zwykle neuropatie

 heterogenna grupa chorób ze wzmożoną syntezą immunoglobulin klas Ig.")
NEUROPATIE PARAPROTEINEMICZNE (MONOKLONALNE, POLIKLONALNE GAMMAPATIE) heterogenna grupa chorób ze wzmożoną syntezą immunoglobulin klas Ig. G, Ig. M, Ig. A ŁAGODNE ZŁOŚLIWE
 • • łagodna gammapatia monoklonalna (MGUS) szpiczak")
POLINEUROPATIE W PRZEBIEGU PARAPROTEINEMII (obecne białko monoklonalne) • • łagodna gammapatia monoklonalna (MGUS) szpiczak pojedyńczy szpiczak mnogi szpiczak osteosklerotyczny! makroglobulinemia Waldenströma choroba Castelmana zespół POEMS ( polineuropatia, organomegalia, endokrynopatia, białko M, zmiany skórne)

• • • P O E M S polineuropatia organomegalia endokrynopatia białko monoklonalne zmiany skórne

Zespół POEMS • Objawy wielonarządowe+ neuropatia uwzględnienie zespołu POEMS w diagnostyce różnicowej • Rozpoznanie zespołu POEMS szczegółowa diagnostyka w kierunku szpiczaka osteosklerotycznego: rtg kości płaskich celowana biopsja!



Neuropatie polekowe • statins

ZESPOŁY PN Z OBWODOWEGO UKŁADU NERWOWEGO PODOSTRA CZUCIOWA NEURONOPATIA OSTRA NEUROPATIA CZUCIOWO-RUCHOWA ZESPÓŁ GUILLAIN BARRE ZAPALENIE SPLOTU BARKOWEGO PODOSTRE I PRZEWLEKŁE NEUROPATIE CZUCIOWO-RUCHOWE NEUROPATIE W WYNIKU ZAPALENIA NACZYŃ (VASCULITIS) NEUROPATIE AUTONOMICZNE PRZEWLEKŁA RZEKOMA NIEDROŻNOŚĆ ŻOŁĄDKA I JELIT OSTRA NIEWYDOLNOŚĆ UKŁADU AUTONOMICZNEGO (PANDYSAUTONOMIA) zespoły klasyczne przeciwciała onkoneuronalne tylko w niektórych guzach przeciwciała onkoneuronalne nieznane
- Slides: 69