In the name of GOD Case presentation Proximal












 LMN (flaccidity, areflexia,")


. l Pseudohypertrophy (infiltration of fat, connective")





 l LDH 647 l ESR 15 l")

")




")
 in chronic denervation- reinnervation (instead")

")







")
 l In some patients inheritance was dominant and in others")
 As a group, their prevalence probably approaches 1 per 100,")
 :")
 The diagnosis is often established by exclusion of everything else.")
 l Treatment is supportive. It is aimed primarily at the")

- Slides: 51
In the name of GOD
Case presentation Proximal muscle weakness with hypertrophy of calves
Brief history l l l A 43 -years old right handed married male. He has history of progressive weakness in his lower limbs and difficulty in walking, inability to climbing and downing from stairs, and inability to standing after sitting down since 11 -years PTA with no history of fluctuation of weakness. He has complain of very mild weakness in upper limbs but no history of muscle pain and sensory complains in his body. He have no history of difficulty in swallowing and breathing. Other systems review were unremarkable. No family history and PMH of medical or surgical diseases but history of large calf since childhood.
Brief N/E l l l 3+/5 Proximal weakness (marked in flexor and adductor of hip, extensor and flexor of knee ) in lower limbs and mild weakness in shoulder abduction with marked atrophy of pelvic girdle (gluteus maximums preserved) and thigh muscles with hypertrophy of calf and EDB muscles and prominent Gower’s sign. Lumbosacral lordosis with waddling gait. Distal muscles and MSR was normal (except knee that was undetectable) and no foot drop. Fasciculation and myotonia were not seen. Other exams were unremarkable except skin change due to superficial vein ecstasy in legs.
Some special signs in muscle exam
Atrophy of quadriceps
Calf hypertrophy
Atrophy of quadriceps and hypertrophy of calf
Hypertrophy of EDB
Lumbosacral lordosis
Distal sparing
Normal upper proximal muscle
localization l l l UMN (Spaticity, Babinski’s sign, Clonus, Hyperreflexia ) LMN (flaccidity, areflexia, fasciculation, generalized atrophy ) Radicuoplexopathy and neuropathy (pain, paresthesia, sensory loss, localized weakness, reflex loss) NMJ (ophtalmoplegia, fluctuation no atrophy) Muscular (weakness, atrophy, preserved reflex, special site involvement)
Another ways to approach:
Approach to Calf hypertrophy with lower LG weakness
Calf hypertrophy l True hypertrophy (true muscle enlargement). l Pseudohypertrophy (infiltration of fat, connective tissue and other materials).
Causes of true calf hypertrophy l S 1 radiculopathy. l MC. l PMC. l Neuromyotonia.
Causes of calf pseudohypertrophy l l l l Duchene's and Becker's muscular dystrophy. Sacroglycanopathies. Spinal muscular atrophies, Kennedy's disease. Glycogen storage disorders. Sarcoidosis, cysticercosis, amyloidosis. Hypothyroid myopathy, focal myositis. Muscle tumors, ruptured tendons, or muscle hernias. Limb-girdle muscular dystrophies.
Approach to EDB hypertrophy with lower LG weakness l Chronic peripheral neuropathy. l Anterior horn cell disease. l Chronic myopathy with proximal weakness. l Muscular dystrophies with proximal weakness.
Approach to thigh atrophy with lower LG weakness l l l l DM (diabetic amyotrophy). Femoral neuropathy. IBM. MND. Distal myopaties. Muscular dystrophy with proximal atrophy. LGMD 2 A (posterior thigh atrophy). LGMD 2 G (quadriceps atrophy).
Approach to distal muscle sparing with LG weakness (rule out lower LG weakness with distal muscle involvement) l Myotonic dystrophy. l IBM. l Miyoshi’s myopathy. l Welander’s myopathy. l Markesbery-Griggs-Udd myopathy. l Nonaka’s myopathy. l Laing’s myopathy. l MND.
Routine Lab test CPK 804 (previously >2000) l LDH 647 l ESR 15 l AST 43 l AST 78 l CRP NEG l VDRL NEG l
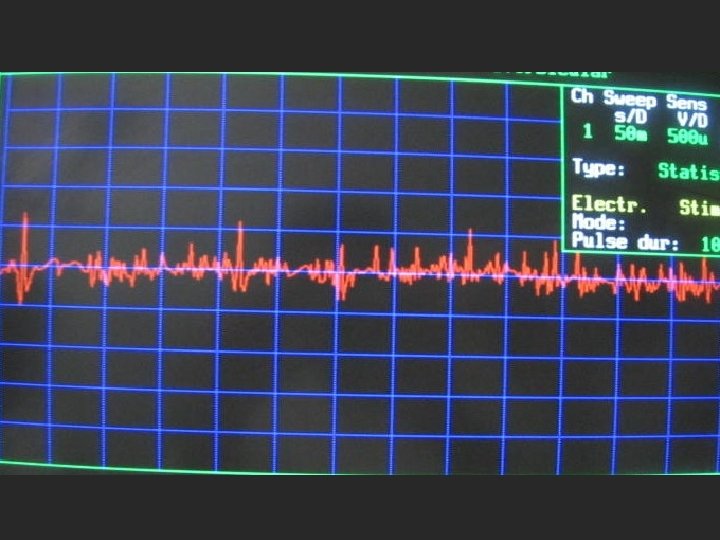
EMG-NCV FINDING l NCV STUDY WAS NORMAL. l EMG: MYOPATHIC CHANGES ( POLYPHASIC MOTOR UNIT WITH REDUCED AMPLITUDE ).
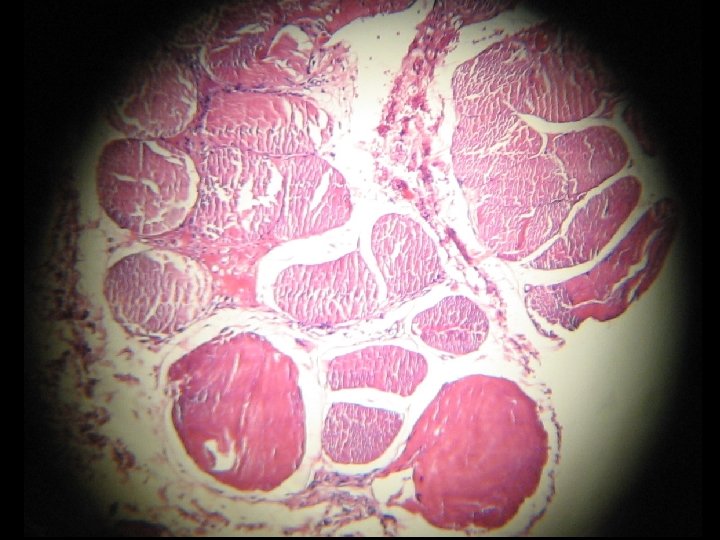
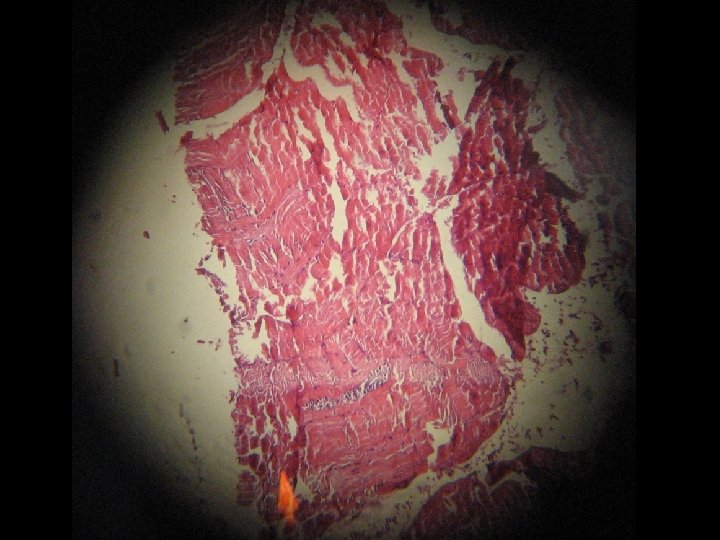
MUSCLE BIOPSY AND PATHOLOGICAL FINDING l l l Fiber size variability. Fiber degeneration (necrosis) and regeneration. Fiber splitting. Fiber atrophy and hyaline changes. Mild lymphocytic infiltration (privascular and endomesial). Perimesial fibrosis and replacing with fat tissue. These findings are in favor of muscular dystrophy.
Types of muscle fibers Based on ATPase staining at PH 9. 2 l Type I : slow-twitch or white-fibers, ability to perform tonic contractions and high in myoglobin, oxidative enzyme, and mitochondria (oxidative fibers). l Type II (two-thirds of muscle fibers) : fasttwitch or red-fibers, ability to perform rapid phasic contractions and high in glycoltic enzyme (glycolytic fibers).
Types of muscle fibers l These fibers are distributed randomly across the muscle, giving rise to the checkerboard or mosaic pattern of alternating light and dark fibers. l Subsets of type II fibers (type IIA have more oxidative metabolism than type IIB, type IIC is present in fetal muscle).
Types of muscle fibers l Since the muscle fibers belonging to one motor unit innervated by the same anterior horn cell are uniform in type (all fibers of single unit are of the same type). l Furthermore, there are fast and slow motor neuron (the motor neuron determines fiber types).
Normal muscle biopsy (ATPase staining at PH 9. 2 )
Muscle biopsy (ATPase staining at PH 9. 2 ) in chronic denervation- reinnervation (instead of mosaic pattern fibers clumped with together, one group next to other
Type I fiber atrophy Nemaline CM. l Central nuclear CM. l Congenital fiber type disproportion CM. l Myotonic dystrophy. l RA. l
Type I fiber atrophy (Myotonic dystrophy)
Type II fiber atrophy l l l l l Disuse atrophy. Pyramidal tract disease. CVD, RA. MR, Cachectic atrophy. Neurodegenerative diseases. Hyperthyroid myopathy. Steroid myopathy. Statins myopathy. Brody disease (stiffness with exercise and deficiency of calcium-activated ATPase). Polymyalgia rheumatica.
Type II fiber atrophy
Fiber type predominance l The name fiber type predominance has been given to a change in the relative numbers of a particular fiber type. l Changes in the proportion of fibers in the biopsy are quite separate from changes in the fiber size.
Type 1 fiber predominance l Type 1 fiber predominance is a normal finding in the gastrocnemius and deltoid muscles. l It is also the hallmark of congenital myopathies and many early-onset dystrophies.
Nemaline CM
Type 2 fiber predominance l Type 2 fiber predominance is seen in the lateral head of the quadriceps muscle. l Type 2 predominance is seen occasionally in juvenile spinal muscular atrophy and motor neuron disease.
Type 2 fiber predominance in chronic denervation- reinnervation
FINAL DIAGNOSIS Limb-girdle muscular dystrophy (LGMD)
Limb-girdle muscular dystrophies (LGMD) l In some patients inheritance was dominant and in others recessive. l LGMD transmitted by autosomal dominant inheritance are designated LGMD type 1 and those transmitted by autosomal recessive inheritances designated LGMD type 2. l The autosomal recessive LGMD are more common than the autosomal dominant LGMD.
Limb-girdle muscular dystrophies (LGMD) As a group, their prevalence probably approaches 1 per 100, 000. l Some had more hip than shoulder weakness and others had the reverse. l The onset of illness could be mild in late life; other patients had severe and early onset. l
Characteristics of some LGMDs l l l l LGMD 1 A (myotilin deficiency) : pharyngeal and facial muscle involvement, 2 th-3 th decade. LGMD 1 B (laminin A/C deficiency) : humeral-peroneal weakness, early contracture, cardiomyopathy and arrhythmia, like to EDMD. LGMD 1 C (caveolin-3 deficiency) : early onset, exertional myalgia, muscle rippling. LGMD 2 A (calpain-3 deficiency) : early onset, winging of scapula, rectus abdominis, posterior thigh. LGMD 2 B (dysferlin deficiency) : early adult onset, like to Miyoshi’s myopathy and missed with polymyositis. LGMD 2 C, D, E, F (sacroglycanopathies) ; child to adult onset, CPK>1000, facial and cardiac sparing, trunk and limb involvement, calf hypertrophy. LGMD 2 G, H, I (sever cardiomyopathy in LGMD 2 I )
Limb-girdle muscular dystrophies (LGMD) The diagnosis is often established by exclusion of everything else. l The most helpful test is the muscle biopsy, which shows dystrophic changes, separating limb girdle dystrophy from other (inflammatory) myopathies, and from denervating diseases. l Immunohistochemical analysis of dystrophic muscle will further clarify the diagnosis, followed by genetic analysis of the sarcoglycan genes. l
Limb-girdle muscular dystrophies (LGMD) l Treatment is supportive. It is aimed primarily at the prevention of contractures, which may result in substantial disability. A passive stretching physical therapy program should be instituted early. Later in the course of the disease, cardiorespiratory status should be monitored.