AME VARIANTE DRA VERNICA SEZ NEURLOGA INFANTIL DRA



 se refiere, en su forma mas común, a")



 Disminución movimientos fetales Subtipo AME 1 Debilidad Hipotonía")


: 967– 973.")






 AME 1 con debilidad severa, tórax en campana,")










 Gen UBA 1 Enzima UBA 1")
 Gen UBA 1 Mutación missense Complejo")


 Aumento concentraciones calcio intracelular AME")




 •")


















- Slides: 62
AME VARIANTE DRA. VERÓNICA SÁEZ NEURÓLOGA INFANTIL DRA. ISADORA RUIZ RESIDENTE NEUROLOGÍA INFANTIL
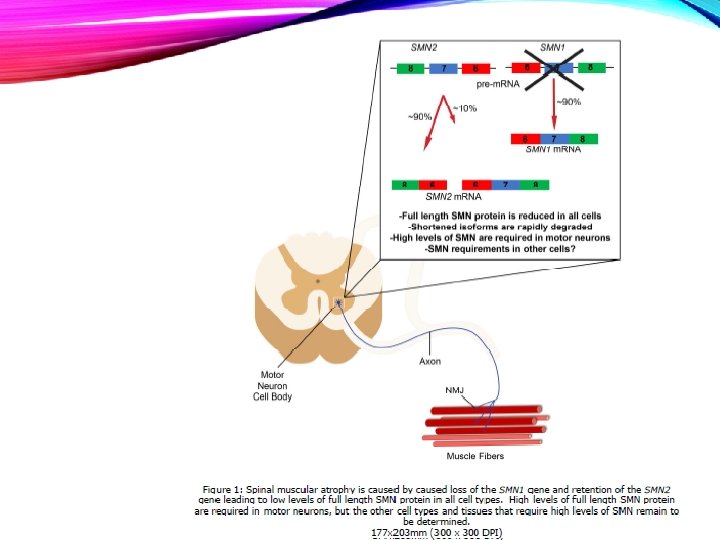
INTRODUCCIÓN • Grupo de trastornos hereditarios caracterizados por degeneración de las células del asta anterior de la médula espinal y ocasionalmente con compromiso de núcleos del tronco (PC: V- XII) SIN COMPROMISO SENSITIVO DEBILIDAD MUSCULAR SIMÉTRICA AUSENCIA SIGNOS PIRAMIDALES • Primera causa genética de muerte en la infancia. • Sin tratamiento modificador de la enfermedad disponible. Emery, A. E. H. The Nosology of the Spinal Muscular Atrophies. J. Med. Genet. 1971, 8, 481– 495.
INTRODUCCIÓN • Fue considerada como una condición genética de herencia exclusivamente autosómica recesiva. • Clasificada en 4 tipos basado en la edad de inicio (Harding y Thomas, 1980) • Gen causal SMN 1 (Le febvre et al, 1995). • Mapeada en el cromosoma 5 (5 q 13). • Proteína de supervivencia de la motoneurona degeneración MN • 4% no asociados a mutación del Cr. 5 q 13
DEFINICIONES La atrofia muscular espinal (AME) se refiere, en su forma mas común, a las causadas por la mutación del gen SMN 1 denominada AME- 5 q o AMErelacionado a supervivencia de MN. AME variante o AME no-5 q son aquellas formas no relacionadas a la mutación de SMN 1. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular trophy determining gene. Cell 1995; 80: 155 -165.
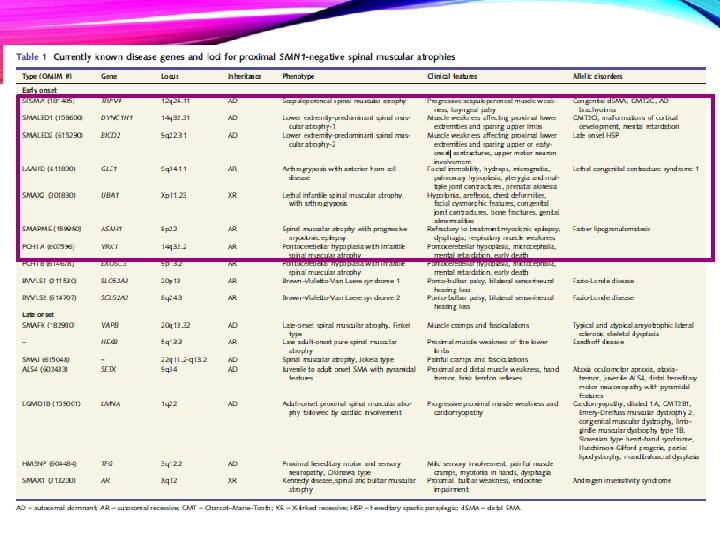
PROGRESOS • Importantes avances en el entendimiento de las bases genéticas y moleculares del AME. • Desde el 2011, se han identificado 16 genes NO-5 q • En total, 33 genes causantes se han identificado a la fecha.
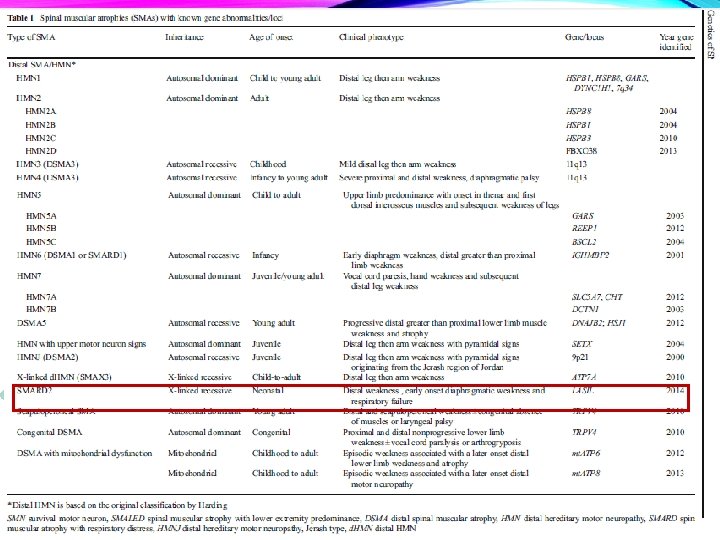
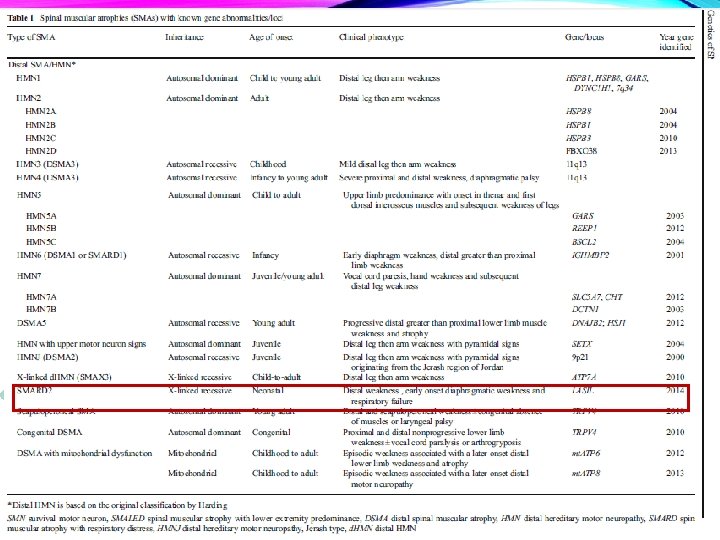
CLASIFICACIÓN AME tipo II AME- 5 q AME tipo III AME tipo IV AME NO-5 q (Variante) PROXIMALES AD AR DISTALES LIG-X
FISIOPATOLOGÍA Neurotherapeutics. DOI 10. 1007/s 13311 -014 -0314 -x
AME DE INICIO ANTENATAL (AME 0) Disminución movimientos fetales Subtipo AME 1 Debilidad Hipotonía marcada al nacer Arreflexia Falla respiratoria por hipoplasia pulmonar Diplegia facial Oftalmoplegia Micrognatia Contracturas proximales y distales La artrogriposis congénita múltiple se ha observado en pacientes con delecciones de SMN 1 Sarnat HB, Trevenen CL (2007). Motor neuron degeneration
• Pacientes con AME a menudo tienen osteopenia severa y experimentan fracturas recurrentes asociadas a un mínimo trauma. • Subgrupo de infantes severamente afectados presenta fracturas óseas congénitas. • Reporte de casos de pacientes que presentan hipercalcemia e hipercalciuria, presuntamente debido a las alteraciones de remodelado óseo secundario a la reducida actividad muscular. • Densidad mineral osea parece disminuir en forma notable al aumentar la edad, sin una relación directa definitiva con un cambio sustancial en la función motora.
MODELO MURINO DE AME • 82% IDENTICOS SMN 1 • MUERTE CELULAR MASIVA EN DESARROLLO • RESCATE DE LETALIDAD • SEMEJANTE A AME SMN 1 -/- J Child Neurol. 2007 August ; 22(8): 967– 973. doi: 10. 1177/0883073807305664 + SMN 2 Degeneración de la cola Vértebras caudales < número tamaño y calidad ósea Fracturas óseas Disminución de área ósea total contenido mineral y densidad ósea
ANORMALIDADES ESQUELÉTICAS EN RATONES AME J Child Neurol. 2007 August ; 22(8): 967– 973. doi: 10. 1177/0883073807305664
ANORMALIDADES ESQUELÉTICAS EN HUMANOS Jourmal of Child Neurology/Volume 17, Number 9, September 2002. Severe spinal Muscular Atrophy variant Associated with congenital Bone Fractures. J Child Neurol. 2007 August ; 22(8): 967– 973. doi: 10. 1177/0883073807305664
• PHA. RNT 40 semanas • Insuficiencia respiratoria al nacer • Micrognatia, paladar ojival. Diplegia facial. Fasciculaciones lengua. Succión deficiente. Hipotonía severa, trofismo disminuido, debilidad severa y generalizada. ROT ausentes. Contracturas múltiples. Fracturas óseas • EMG: signos de denervación extensa en todos los músculos examinados • Delección homocigota de SMN 1 y presencia de SMN 2 (una copia) La presencia de características atípicas durante el periodo neonatal no descarta el diagnóstico de AME 5 -q
ROL FUNCIONAL DEL SMN EN DESARROLLO ESQUELÉTICO Y REMODELACIÓN ÓSEA J Child Neurol. 2007 August ; 22(8): 967– 973. doi: 10. 1177/0883073807305664
AME VARIANTE
AME VARIANTE. AME NO-5 Q O AME “PLUS” • Formas raras, clínica y genéticamente heterogéneas. AME • Compromiso: debilidad y atrofia • El diagnóstico diferencial incluye: - AME escápulo-humeral - AME infantil con artrogriposis ligado a X (XL-SMA) - AME con hipoplasia pontocerebelar (SMA-PCH/PCH 1) - AME con distress respiratorio (SMARD) - AME con epilepsia mioclónica progresiva AME 5 q AME NO-5 q (Variante) PROXIMALES Ad AR DISTALES LIG-X
CARACTERÍSTICAS CLÍNICAS • Espectro clínico y genético heterogéneo desde muerte neonatal hasta vida en adultez normal con debilidad muscular leve. • Importancia de historia clínica detallada y examen neurológico exhaustivo. • Rasgos de herencia no siempre presente, debido a mutaciones de novo esporádicas, por lo cual es preferible orientar diagnóstico en función de la edad de inicio y compromiso predominante.
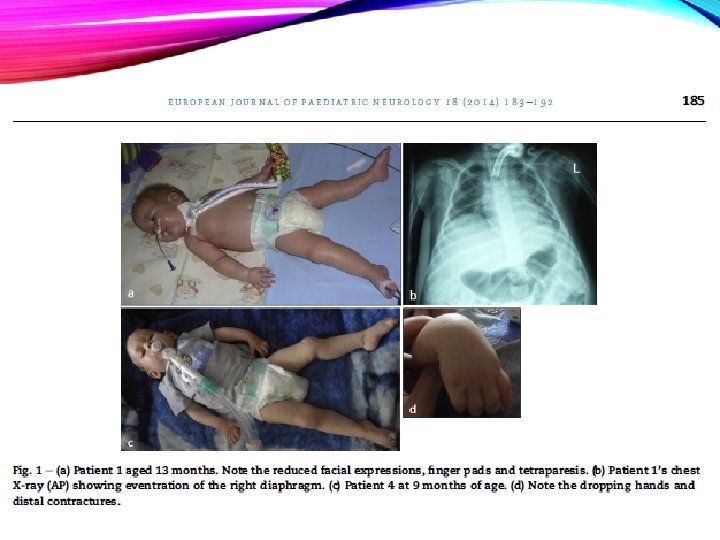
AMPLIO ESPECTRO DE MANIFESTACIONES CLÍNICAS a) AME 1 con debilidad severa, tórax en campana, I. respiratoria b) Rx. toracolumbar de paciente AME 2 con escoliosis TL c) d) HNM 5 C por mutación BSCL 2: atrofia distal EEII e) HNM 5 C por mutación BSCL 2: debilidad y atrofia de manos con dedos contracturados g) Pie cavo h) Rx tórax en paciente con SMARD 1 que muestra eventración de hemidiafragma derecho
ELECTROFISIOLOGÍA ESTUDIO ELECTROFISIOLÓGICO NO ES SIEMPRE TÍPICO AL INICIO DE LA ENFERMEDAD Hausmanowa-Petrusewicz, I. , and Karwanacuteska, A. (1986) Muscle Nerve, 9, 37 -46.
GENES INVOLUCRADOS
FISIOPATOLOGÍA
AME VARIANTE PROXIMAL DE INICIO PRECOZ
AME VARIANTE PROXIMAL • Debilidad muscular simétrica de predominio proximal, con mayor compromiso de EEII generalmente. (D´Amico et al, 2011) • ROT ausentes • Estático a rápidamente progresivo hasta requerimiento de ventilación mecánica. • Sensitivo, bulbares y cognitivo preservado habitualmente • Deformidades y contracturas Maystadt I, Rezsohazy R, Barkats M, et al. The nuclear factor kappa. B-activator gene PLEKHG 5 is mutated in a form of autosomal recessive lowermotor neuron diseasewith childhood onset. Am J Hum Genet 2007; 81: 67 -76.
AME PROXIMAL DE INICIO PRECOZ CON ARTROGRIPOSIS LETAL CON ENFERMEDAD DE CÉLULA DEL ASTA ANTERIOR (AR) AME INFANTIL LETAL CON ARTROGRIPOSIS (LIG-X) • De las formas severas de enfermedad de motoneurona. • La muerte ocurre en forma temprana en periodo neonatal, debido a falla respiratoria • EMG y biopsia muscular compatible con pérdida células del asta anterior. • Patología: ausencia de neuronas del asta anterior, atrofia severa del ME ventral e hipoplasia o incluso, ausencia de músculos. Greenberg F, Fenolio KR, Hejtmancik JF, Armstrong D, Willis JK, Shapira E, et al. X-linked infantile spinal muscular atrophy. Am J Dis Child 1988; 142: 217– 9.
ARTROGRIPOSIS LETAL CON ENFERMEDAD DE CÉLULA DEL ASTA ANTERIOR INMOVILIDAD FETAL LAAHD HIDROPS ANOMALÍAS FACIALES: MICROGNATIA. CUELLO CORTO HIPOPLASIA PULMONAR PTERIGIO CONTRACTURAS MULTIPLES FRACTURAS ÓSEAS CONGÉNITAS AUTOSÓMICO RECESIVO GEN GLE 1, Cr. 9 OMMIM 611890 LAAHD
ARTROGRIPOSIS LETAL CON ENFERMEDAD DE CÉLULA DEL ASTA ANTERIOR Gen GLE 1 Neuroporina (forma de disco) Complejos de poro nuclear Exporta el m. RNA desde el núcleo al citoplasma
ARTROGRIPOSIS LETAL CON ENFERMEDAD DE CÉLULA DEL ASTA ANTERIOR Gen GLE 1 Mutación homocigota Partículas desordenadas y malformadas Desregulación de la remodelación del RNAm Enlentecimiento del transporte del RNAm
PHA AME INFANTIL LETAL CON ARTROGRIPOSIS LIGADO-X HIPOTONÍA SEVERA SMAX-2 ARREFLEXIA ARTROGRIPOSIS LIGADO A X MUTACIÓN GEN UBA 1, Cr. X OMIM 301830 SMAX-2 DEFORMIDADES TORÁCICAS DISMORFIAS FACIALES FRACTURAS CONGÉNITAS SIMILAR AME 1 Greenberg F, Fenolio KR, Hejtmancik JF, Armstrong D, Willis JK, Shapira E, et al. X-linked infantile spinal muscular atrophy. Am J Dis Child 1988; 142: 217– 9.
ATROFIA MUSCULAR ESPINAL LIGADA A X (SMAX 2) Gen UBA 1 Enzima UBA 1 Forma complejo con gigaxonina (GAN) Activación y conjugación de proteínas tipo ubiquitina Estructura axonal y mantenimiento neuronal Controla degradación de proteína asociada a microtúbulos 1 B (MAP 1 B)
ATROFIA MUSCULAR ESPINAL LIGADA A X (SMAX 2) Gen UBA 1 Mutación missense Complejo GAN + UBA 1 alterado Menor degradación MAP 1 B (sobreexpresión de MAP 1 B) Compromiso de la supervivencia neuronal
AME ESCAPULO-HUMERAL DE INICIO TEMPRANO PRESENTACIÓN CONGÉNITA O INFANCIA AME ESCAPULO-HUMERAL DE INICIO TEMPRANO ATROFIA ESCAPULOHUMERAL PROGRESIVA DEBILIDAD PROXIMAL EN EESS Y DISTAL EN EEII PARÁLISIS LARÍNGEA Y VOZ RONCA AR MUTACIÓN GEN TRPV 4, Cr. 12 OMIM 181405 SPSMA RDSM COGNITIVO NORMAL BIOPSIA: ATROFIA DE FIBRAS AGRUPADAS, PROCESO NEUROGÉNICO De. Long R, Siddique T. A large New England kindred with autosomal dominant neurogenic scapuloperoneal amyotrophy with unique features. Arch Neurol 1992; 49: 905– 8.
GENES IMPLICADOS TRPV 4 Canal de calcio no selectivo (Receptor de potencial transitorio de canal catiónico) Rol a nivel de la señalización neuronal. Deng HX, Klein CJ, Yan J, Shi Y, Wu Y, Fecto F, et al. Scapuloperoneal spinal muscular atrophy and CMT 2 C are allelic disorders caused by alterations in TRPV 4. Nat Genet 2010; 42: 165– 69.
AME escapulohumeral TRPV 4 TRASTORNOS ALÉLICOS Mutación missense (AD) Aumento concentraciones calcio intracelular AME distal Neuropatía hereditaria sensitivo motora 2 C Reduce la superficie de expresión del canal Formación de agregados citoplasmáticos Pérdida de la función del canal Deng HX, Klein CJ, Yan J, Shi Y, Wu Y, Fecto F, et al. Scapuloperoneal spinal muscular atrophy and CMT 2 C are allelic disorders caused by alterations in TRPV 4. Nat Genet 2010; 42: 165– 69.
Efectos fenotípicos variados debido a diferentes interacciones reguladoras entre proteínas Reduced penetrance in hereditary motor neuropathy caused by TRPV 4 Arg 269 Cys mutation. J Neurol (2011) 258; 1413 -1421
AME DE PREDOMINIO EEII ATROFIA Y DEBILIDAD PROXIMAL >EEII AME DE PREDOMINIO EEII DEBILIDAD LEVE O AUSENTE DE EESS INICIO TEMPRANO CURSO VARIABLE DEAMBULACIÓN HASTA 6° DÉCADA DEFORMIDADES ESQUELÉTICAS AUTOSÓMICO DOMINANTE GEN DYNC 1 H 1, Cr. 14 OMIM 158600 SMA-LED HIPERLORDOSIS LUMBAR ESCAPULA ALADA DISLOCACIÓN CADERAS VCN NORMAL EMG: DENERVACIÓN Y REINERVACIÓN CRÓNICA BIOPSIA: DENERVACIÓN Y REINERVACIÓN CRÓNICA CONTRACTURAS EEII
GENES IMPLICADOS • Mutación en la cadena pesada de Dineina citoplasmática. DYNC 1 H 1 (proteína asociada a microtúbulos)-> Trasporte axonal retrogrado - 4 mutaciones sentido erróneo heterocigotas asociadas a AMELED • Gen BICD 2 • Mutación puntual de gen de DYMC 1 H 1 también se ha encontrado en familias con CMT 20 • Mutaciones del DYMC 1 H 1: - DI - Alteraciones en la migración de corteza - Paraplejia espástica hereditaria
AME CON EPILEPSIA MIOCLÓNICA PROGRESIVA INICIO ENTRE 3 -5 AÑOS DEBILIDAD PROGRESIVA DE EESS Y EEII EPILEPSIA MIOCLÓNICA DE APARICIÓN TARDÍA AUTOSÓMICO RECESIVO GEN ASAH 1, Cr. 8 OMIM 159950 SMAPME EPILEPSIA DE CURSO REFRACTARIO DISFAGIA COMPROMISO RESPIRATORIO INFECCIONES RESPIRATORAS DISCAPACIDAD SEVERA MUERTE < 20 AÑOS Zhou J, Tawk M, Tiziano FD, Veillet J, Bayes M, Nolent F, et al.
GENES INVOLUCRADOS • Mutación en el gen que codifica N-acylsphingosine amidohydrolase (ASAH 1) • ASAH 1 es una enzima lisosomal que degrada la ceramida en esfingosina y ácidos grasos libres. • Mutación: no influye en la transcripción o expresión de la proteína. Reduce actividad a 30%, • Los pacientes con Enfermedad de Farber presentan actividad aún menor. • Se propone que la mayor actividad residual enzimática en pacientes con SMA-PME es responsable del fenotipo de inicio tardío, restringido a las MN y otras áreas del SNC, en comparación con la Enfermedad multisistémica de Farber de inicio temprano
AME CON HIPOPLASIA PONTOCEREBELAR RDSM MICROCEFALIA HIPOTONÍA SEVERA AL NACER AUTOSÓMICO RECESIVO GEN EXOSC 3, Cr. 9 OMIM 614678 PCH • Grupo de trastornos neurodegenerativos que afectan el desarrollo y función del tronco cerebral y cerebelo DEBILIDAD TRONCO Y EE ARREFLEXIA • Trastorno motor central y periférico con degeneración de MN y muerte precoz por insuficiencia respiratoria. SIGNOS CEREBELOSOS • Patología: degeneración de células del asta anterior y pérdida de células de Purkinje y granulares con gliosis en cerebelo CONTRACTURAS EMG: COMPROMISO NEUROGÉNICO SIN SENSITIVO • Pérdida neuronal además en: ME, G. de la base, tronco degeneración neuronal amplia. Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, et al.
AME CON HIPOPLASIA PONTOCEREBELAR
GENES IMPLICADOS • AR • Mutación EXOSC 3: mayor causada de PCH-1 de severidad variable. • EXOSC 3: componente central del complejo exosoma de RNA (COMPLEJO MULTIPROTEICO CAPAZ DE DEGRADAR DIVERSOS TIPOS DE RNA) • Primera enfermedad asociada con esta disfunción Rudnick-Schöneborn, S. ; Senderek, J. ; Jen, J. J. , et al. Pontocerebellar
AME DISTAL
AME CON DISTRESS RESPIRATORIO TIPO 1 DISTRESS RESPIRATORIO PARÁLISIS DIAFRAGMÁTICA LUEGO: DEBILIDAD DISTAL ATROFIA MUSCULAR Y DEBILIDAD PROGRESIÓN HASTA TETRAPLEGIA DISFUNCIÓN AUTONÓMICA COGNITIVO NORMAL Antes de 1° año de edad AUTOSÓMICO RECESIVO GEN IGHMBP 2. Cr. 11 OMIM 604320 SMARD O NEURONOPATÍA HEREDITARIA MOTORA TIPO VI (DSMA 1)
GENES IMPLICADOS • Mutación del gen IGHMBP 2. Cromosoma 11 q 13. 3 • Helicasa dependiente de ATP: separa la doble hebra de DNA y RNA • Mecanismo desconocido: ¿compromiso metabolismo RNA? Lim SC, Bowler MW, Lai TF, Song H. The IGHMBP 2 helicase
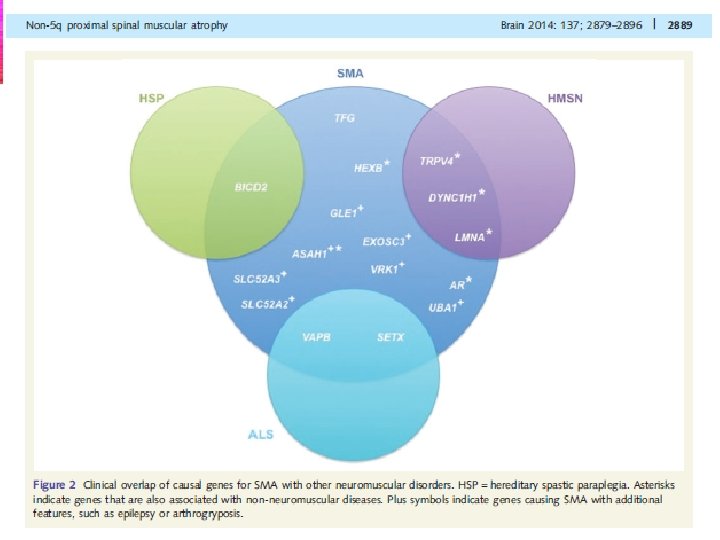
NOVEDADES EN FISIOPATOLOGÍA DE AME • AME proximal no-5 q son trastornos raros que representan un desafío para su diagnóstico y manejo. • Este grupo heterogéneo demuestra el superposición con otros trastornos Neuromusculares como: HMSN, paraplegia espástica hereditaria y ELA • Los genes causantes de AME son en su mayoría expresados en forma ubicua y sus defectos moleculares pueden afectar otros tejidos causando por ejemplo diversas Laminopatias (LMNA), displasias esqueléticas (TRPV 4) y malformaciones del desarrollo cortical (DYNC 1 H 1)
CONCLUSIONES • A pesar del numero creciente de genes identificados a abierto el abanico de mecanismos fisiopatológicos no hay un mecanismo de enfermedad identificado. Vías comunes: Defectos metabolismo Splicing RNA Trasporte axonal Desarrollo y conectividad VULNERABILIDAD MOTONEURONA
Brain 2014: 137; 2879– 2896
DESAFÍOS • Hace 20 años: Casos de AME no- q diagnóstico anecdótico • Hoy existe un número creciente de condiciones asociadas a esta categoría clínica con un gran espectro de genes identificados gracias al desarrollo de tecnología de secuenciación de nueva generación • Desentrañar la etiología de las diferentes formas de SMA requerirán una comprensión en profundidad de la función de las proteínas mutadas en complejas funciones celulares Objetivo importante de la investigación futura. El espectro fenotípico asociado con muchos de los genes en AME es demasiado amplio o no conoce lo suficiente como para identificar el subtipo correspondiente en muchos casos clínicos. Además, las pruebas genéticas se ofrece por sólo un puñado de laboratorios internacionales dispersos por todo el mundo
MANEJO MULTIDISCIPLINARIO KINESIOTERAPIA ESTADO FUNCIONAL DEL PACIENTE FISIATRÍA MANEJO RESPIRATORIO AVNI APOYO EMOCIONAL NUTRICIÓN MANEJO CONTRACTURAS OBJETIVOS DE PRÁCTICA MÉDICA SEGÚN: EXPECTATIVAS DE LA FAMILIA
¿ES ENFERMEDAD DE MOTONEURONA? SI NO Neuromuscular: - Distrofia muscular - Distrofia miotónica - Síndromes miasténicos - Miopatías metabólicas - Neuropatia hipomielinizante congénita No neuromusculares: - EHI - Sepsis neonatal - Metabólico Buscar: - Artrogriposis - Epilepsia mioclónica - Hipoacusia SN - Hipoplasia pontocerebelar AME NO- 5 q Gen SMN 1 S 95%- E 100% OTROS GENES No-q PROXIMAL AD AME 5 q DISTAL AR LIG-X
CASO CLÍNICO
DISMINUCIÓN MOVIMIENTOS FETALES INMOVILIDAD FETAL DEBILIDAD ANOMALÍAS FACIALES: MICROGNATIA. CUELLO CORTO HIPOPLASIA PULMONAR PTERIGIO MICROGNATIA CONTRACTURAS MULTIPLES FRACTURAS ÓSEAS CONTRACTURAS PROXIMALES Y DISTALES FRACTURAS ÓSEAS CONGÉNITAS ARREFLEXIA SMAX-2 DIPLEGIA FACIAL LAAHD AME 0 FALLA RESPIRATORIA POR HIPOPLASIA PULMONAR HIPOTONÍA SEVERA HIDROPS HIPOTONÍA MARCADA AL NACER ARREFLEXIA PHA ARTROGRIPOSIS DEFORMIDADES TORÁCICAS DISMORFIAS FACIALES FRACTURAS CONGÉNITAS SIMILAR AME 1
DISTRESS RESPIRATORIO SMARD PARÁLISIS DIAFRAGMÁTICA LUEGO: DEBILIDAD DISTAL ATROFIA MUSCULAR Y DEBILIDAD PROGRESIÓN HASTA TETRAPLEGIA DISFUNCIÓN AUTONÓMICA COGNITIVO NORMAL
DIAGNÓSTICOS • SINDROMÁTICO 1. RDSM GLOBAL 2. SINDROME HIPOTÓNICO MIXTO 3. ARTROGRIPOSIS • LOCALIZATORIO 1. 2° MOTONEURONA V/S MULTISISTÉMICO? • ETIOLÓGICO 1. ATROFIA MUSCULAR ESPINAL ASOCIADA A SMN 1 (AME 0) V/S ATROFIA MUSCULAR ESPINAL VARIANTE (SMAX-2) • GENERALES 1. FRACTURA DE HÚMERO CONGÉNITA BILATERAL 2. CRIPTOQUIDEA
• GRACIAS