TRASLOCACION FARMACOLOGIA BASICA TRANSFERENCIA DE FARMACOS TRANSFERENCIA DE

TRASLOCACION FARMACOLOGIA BASICA TRANSFERENCIA DE FARMACOS
 de fármacos a través de barreras membranales puede")
TRANSFERENCIA DE FARMARMACOS La transferencia (translocación) de fármacos a través de barreras membranales puede realizarse por: filtración, difusión, transporte activo, pinocitosis o fagocitosis (procesos en los que la célula envuelve e introduce moléculas a su interior).

La diferencia de estos procesos depende del tamaño de la droga que se transporte, su solubilidad y la necesidad de acarreadores membranales. Para la filtración y la difusión, la velocidad de transferencia depende también del gradiente de concentración del fármaco en ambos lados de la membrana.

En el caso del transporte activo, una sustancia puede ser introducida al espacio intracelular independientemente de su tamaño o liposolubilidad; sin embargo, en esta situación se requiere de cierta especificidad estructural; recordemos que este transporte activo es un mecanismo saturable y dependiente de energía.

BARRERAS BIOLOGICAS FARMACOLOGIA

BARRERAS BIOLOGICAS Desde el punto de vista farmacocinético las barreras son dispositivos limitantes de los compartimientos. El paso de fármacos a través de las barreras biológicas está condicionado por las características fisicoquímicas de la sustancia.

Paso de medicamentos a través de la Placenta El concepto de que la placenta constituye una barrera para los fármacos es falso, el feto esta expuesto en cierto grado, a todos los fármacos administrados a la madre. La administración de fármacos en el 1 er. Trimestre de gestación puede provocar efectos teratógenos. En cuanto al feto, la distribución de todo tipo de sustancias es a través de los vasos umbilicales formados por tejido placentario.

La placenta es un órgano en sí: tiene una estructura anatómica definida, con capacidad de filtrar y metabolizar sustancias provenientes de la sangre materna (incluyendo fármacos), así como de producir hormonas necesarias para el proceso de la gestación, además, sirve de barrera entre el ambiente de la madre y el feto; y las drogas que atraviesan la barrera placentaria pueden afectar al feto a veces en forma irreversible.
, pasan a través de")
En general: los fármacos liposolubles (morfina, barbitúricos, anestésicos generales), pasan a través de la placenta por difusión pasiva. la glucosa por difusión facilitada; los iones y aminoácidos por transporte activo las proteínas (inmunoglobulinas) por pinocitosis los amonios cuaternarios (relajantes musculares) y las sustancias hidrosolubles de alto peso molecular (mayor de 1000) no atraviesan la barrera placentaria.

La mayoría de los fármacos atraviesan la membrana por difusión simple. Mencionemos los casos especiales del paso de fármacos al sistema nervioso y al feto. Por otra parte, dado que el p. H de la sangre fetal es ligeramente inferior al de la sangre materna, tienden a acumularse en el feto los medicamentos de carácter básico.

En el caso del cerebro y médula espinal, muchas sustancias pasan de la sangre al líquido cefalorraquídeo (LCR) de los ventrículos cerebrales. El LCR se forma cuando la sangre pasa a través de los plexos coroideos donde células especializadas filtran y cambian su composición.

El LCR transporta sustancias alimenticias, hormonas o productos de desecho a los sitios más profundos del SNC, allí donde los vasos sanguíneos son demasiado pequeños o insuficientes para mantener la función de esas estructuras.

ESTRUCTURA GENERAL DE LA MEMBRANA CELULAR FARMACOLOGIA

Membrana celular La membrana está constituída de lípidos y proteínas. La parte lipídica de la membrana está formada por una película bimolecular que le da estructura y constituye una barrera que impide el paso de substancias hidrosolubles.
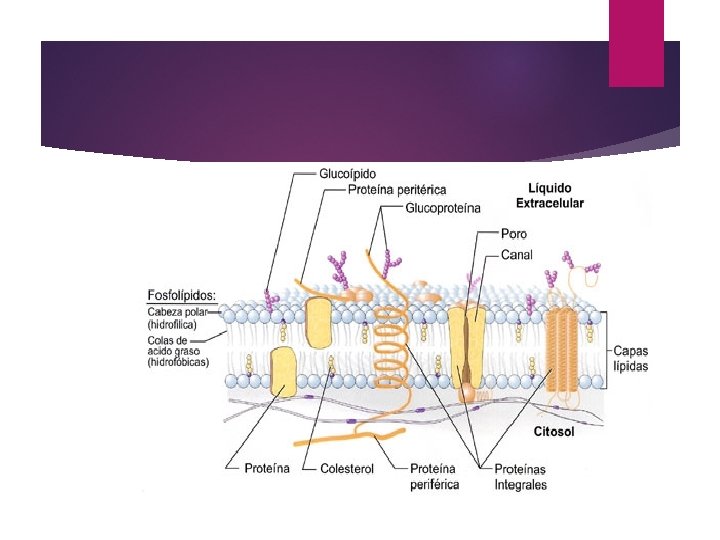

Las proteínas de la membrana están suspendidas en forma individual o en grupos dentro de la estructura lipídica, formando los canales por los cuales entran a las células, en forma selectiva, ciertas substancias. La selectividad de los canales de proteínas le permite a la célula controlar la salida y entrada de substancias así como los transportes entre compartimentos celulares.

Las proteínas de la membrana no solo hacen que el transporte a través de ella selectivo, sino que también son capaces de llevar a cabo transporte activo (transferencia en contra del gradiente de concentración).

Las demás funciones de la membrana, como son el reconocimiento y unión de determinadas substancias en la superficies celular están determinadas también por la parte proteica de la membrana. A estas proteínas se les llaman receptores celulares. Los receptores están conectados a sistemas internos que solo actúan cuando la sustancia se une a la superficie de la membrana.

En la membrana se localizan unas glucoproteínas que identifican a otras células como integrantes de un individuo o como extrañas (inmunoreacción). Las interacciones entre las células que conforman un tejido están basadas en las proteínas de las membranas.
 liposolubilidad (capacidad")
En particular: tamaño o peso molecular; grado de ionización (carga eléctrica) liposolubilidad (capacidad de disolverse en las grasas). Así, una sustancia pequeña, poco ionizada y muy liposoluble atraviesa rápidamente las membranas celulares. Tal es el caso de la mayoría de los anestésicos volátiles, agentes broncodilatadores o solventes orgánicos.

Así, una sustancia pequeña, poco ionizada y muy liposoluble atraviesa rápidamente las membranas celulares. Tal es el caso de la mayoría de los anestésicos volátiles, agentes broncodilatadores o solventes orgánicos.

MECANISMOS GENERALES DE TRANSFERENCIA FARMACOLOGIA

Difusión: El movimiento espontáneo de partículas desde un área de alta concentración a un área de baja concentración

: La incorporación de material")
Endocitosis: (del griego endon = dentro; kytos = célula): La incorporación de material desde el exterior de la célula hacia el interior por la formación, en la membrana plasmática, de una vesícula que rodea al material en manera tal que la célula lo pueda incorporar. Incluye 1) fagocitosis 2) pinocitosis 3) endocitosis mediada por receptor
 engloban")
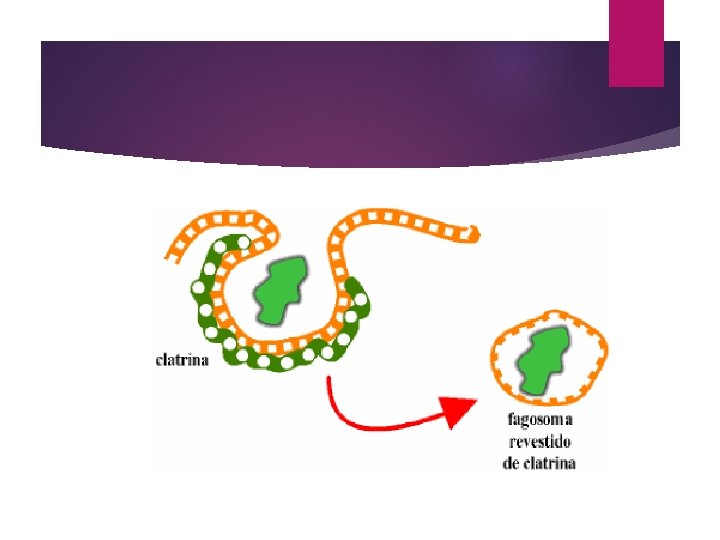
Fagocitosis: Una forma de endocitosis en la cual células sanguíneas (los fagocitos) engloban partículas (entre otras, bacterias); en este proceso, la célula crea una proyecciones de la membrana y el citosol llamadas pseudopodos que rodean la partícula sólida. Una vez rodeada, los pseudopodos se fusionan formando una vesícula alrededor de la partícula llamada vesícula fagocítica o fagosoma.

El material sólido dentro de la vesícula es seguidamente digerido por enzimas liberadas por los lisosomas. Los glóbulos blancos constituyen el ejemplo más notable de células que fagocitan bacterias y otras sustancias extrañas como mecanismo de defensa

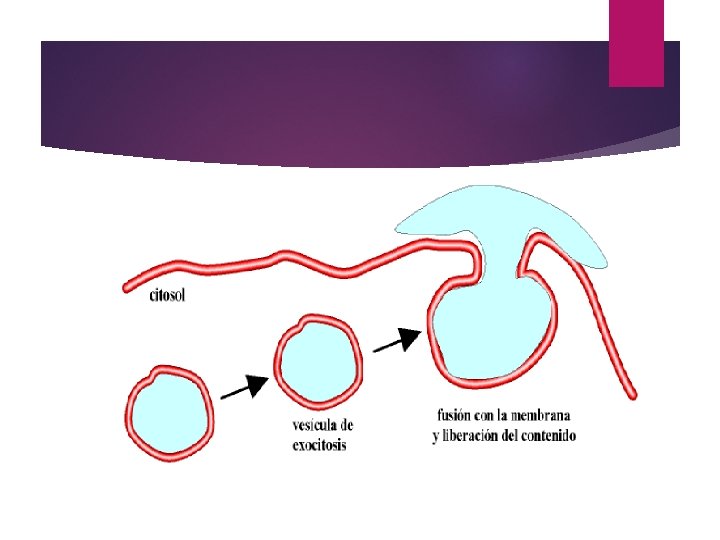
Exocitosis: El proceso en el cual una vesícula primero se fusiona con la membrana plasmática y luego se abre y libera su contenido al exterior.

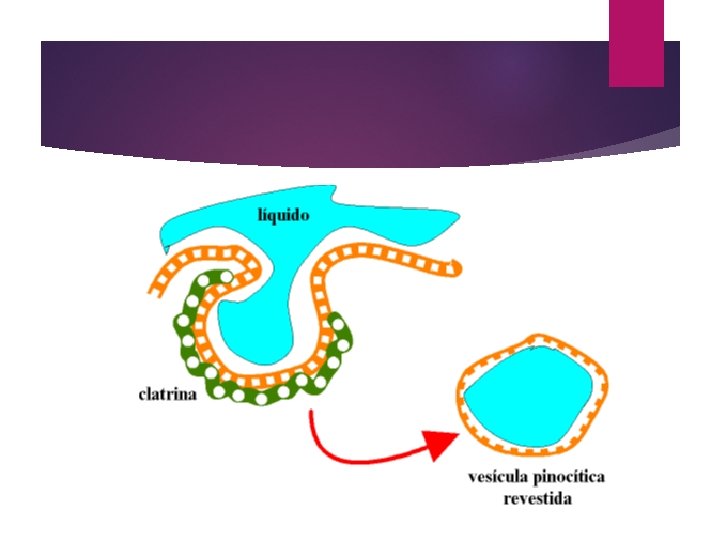
Pinocitosis: en este proceso, la sustancia a transportar es una gotita o vesícula de líquido extracelular. En este caso, no se forman pseudópodos, sino que la membrana se repliega creando una vesícula pinocítica. Una vez que el contenido de la vesícula ha sido procesado, la membrana de la vesícula vuelve a la superficie de la célula.

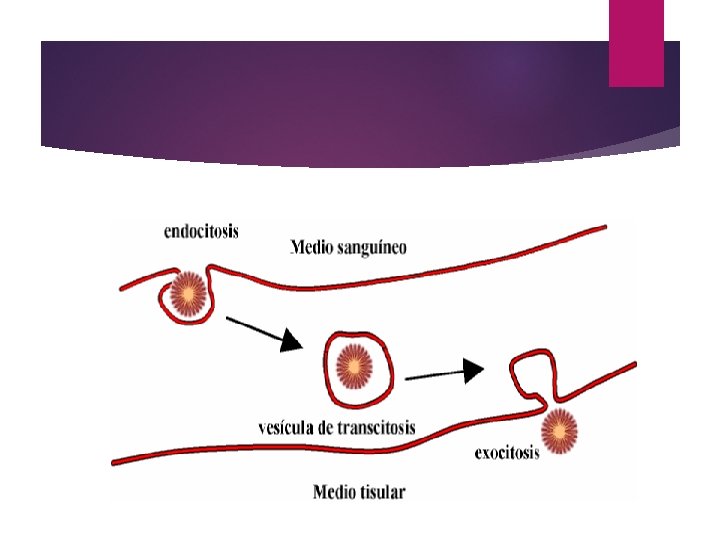
Transcitosis. Es el conjunto de fenómenos que permiten a una sustancia atravesar todo el citoplasma celular desde un polo al otro de la célula. Implica el doble proceso endocitosis-exocitosis. Es propio de células endoteliales que constituyen los capilares sanguineos, transportándose así las sustancias desde el medio sanguineo hasta los tejidos que rodean los capilares.

Transporte activo: Transporte de moléculas contra un gradiente de concentración (de regiones de baja concentración a regiones de alta concentración) con ayuda de proteínas de la membrana celular y energía proveniente del ATP. Transporte pasivo: Difusión a través de la membrana plasmática sin gasto energético por parte de la célula

FACTORES FISICOQUIMICOS DE LAS MOLECULAS DE UN FARMACO QUE INFLUYEN EN LA TRANSLOCACION FARMACOLOGIA

La distribución del fármaco dentro del cuerpo puede variar de acuerdo con el flujo sanguíneo o la vascularización regional de cada tejido u órgano, y la cantidad de droga que cada tejido reciba depende de la concentración del fármaco en la sangre.

A su vez, la magnitud del efecto varía por la velocidad con la que el fármaco penetra al tejido hasta alcanzar niveles suficientes. Para que una sustancia atraviese las membranas celulares es condición esencial que se encuentre en forma libre, es decir, que no esté unida a otras moléculas.

En la sangre, la albúmina representa una proteína con múltiples sitios de unión para fármacos. Mientras éstos se mantengan unidos a la albúmina no podrán abandonar el torrente sanguíneo y, por lo tanto, no llegarán a sus sitios de acción.

Por otra parte, los fármacos, a su vez, competirán con otras moléculas endógenas contenidas en la sangre (p ejem. , hormonas, bilirrubina, vitaminas, iones, etc. ) por los sitios de transporte, consecuencias potencialmente peligrosas de acumulación.

En relación con la distribución del fármaco, una vez que alcanza el espacio intravascular, es necesario tomar en cuenta su volumen aparente de distribución (Vd), o sea, el volumen fluido en el que el fármaco se distribuye, puesto que es un índice de la compartimentalización de la sustancia.

Un fármaco con Vd elevado es una sustancia que se almacena o secuestra en algún compartimiento del organismo, por lo que tendrá un potencial de toxicidad por acumulación. El Vd es diferente entre niños y adultos, y entre sujetos sanos y enfermos.

Así, la distribución de una fármaco determinará en parte la latencia, intensidad y duración de la actividad biológica del fármaco. Existen varios factores que pueden afectar el Vd: la afinidad del fármaco por las moléculas transportadas por la sangre el flujo sanguíneo regional la afinidad por los componentes de los tejidos las barreras especiales (p. ejem. , placenta, cerebro) factores fisiológicos (p. ejem. , ritmos biológicos, p. H, glicemia, etc. ) patológicos (p. ejem. , inflamación o edema, diarrea, fiebre, etc. ) farmacológicos (p. ejem. , interacción con otras sustancias).

El ritmo de absorción y eliminación de un fármaco depende de los procesos citados anteriormente y determina la frecuencia de administración del medicamento. La farmacocinética integral utiliza el concepto de vida media (el tiempo necesario para que la concentración sanguínea del fármaco se reduzca a la mitad).

Cuando se administra un fármaco se trata de establecer una concentración terapéutica en los fluidos biológicos. Esta concentración eficaz es una propiedad característica del fármaco sobre la cual no tenemos control.

Si el nivel de la droga en el suero es insuficiente, la respuesta deseada no ocurre. Si el nivel es más elevado, aparecen signos de toxicidad. Los horarios de dosificación comprenden dos variables: la magnitud de la dosis única y la frecuencia con que se administra (intervalo entre las dosis).

Las fluctuaciones de los niveles séricos que pueden observarse entre las administraciones son determinadas por varios factores: para un ritmo dado de eliminación, mientras más rápida es la absorción, más grande es la fluctuación. Si la absorción es rápida, los niveles sanguíneos se elevan al principio, pero disminuyen también relativamente rápido y viceversa.

Finalmente, es necesario considerar la biodisponibilidad, entendida como la facilidad con la que un fármaco se incorpora a sus sitios de acción; aquí se incluye la presentación farmacéutica en la que se ofrece el medicamento. La absorción no es la misma para una tableta que para una cápsula, que para una preparación de liberación prolongada.

En este último caso, la sustancia activa se halla incluida en una matriz de degradación lenta que va liberando gradualmente el principio activo, y como la dosis que se administra representa varias dosis únicas, existe el peligro potencial de una liberación masiva del fármaco contenido en la preparación y los consecuentes efectos tóxicos por sobredosis.

VIAS DE ADMINISTRACIÓN Y DISTRIBUCIÓN

FIN FARMACOLOGIA
- Slides: 53