pharmacokinetics Department of pharmacology Yang Fangju 2010 3
 2010. 3")



 l The nonionized molecules are usually lipid")
 Active transport l This is a process in which a solute moves")


![10 p. H -pka = [ A- ] [ HA ] When p. H](https://slidetodoc.com/presentation_image_h/85ab75ccd3457ad892e889c14e0a6571/image-10.jpg "10 p. H -pka = [ A- ] [ HA ] When p. H")
 absorption l ①absorption of")












 Renal excretion l Excretion of drugs and metabolites in the urine involves three")
 Excretion by other routes l The metabolites of drugs are also excreted into")

 drug absorption and C-T curve l ① peak concentration (Cmax) l ② peak")

 distribution of the drug and C-T curve l ① one-compartment model l")

 l the volume of distribution (V) relates the amount")

=-ket, t=ln(C/C 0)/-k l when t =")

 drug is cleared in an exponential manner")


 Repetitive")

 if: t = t 1/2, then:")


- Slides: 49
pharmacokinetics Department of pharmacology Yang Fang-ju (杨芳矩) 2010. 3
Section 1 drug process in the body l 1. classification of drug process in the body l It includes absorption, distribution, metabolism, excretion. l Metabolism and excretion are called elimination.
tissue, cells dissociatio n bind Blood plasma absorption Dissociated drug Bound drug excretion metabolite biotransformation Process of drugs in the body
l 2. transportation of drugs across membranes l The absorption, distribution, biotransformation, and excretion of a drug all involve its passage across cell membranes. l Transported model: l ⑴Passive diffusion l It transports along a concentration gradient l ※ It needs not metabolic energy l ※ it is usually determined by its p. Ka, lipidwater partition coefficient, molecular weigh, and p. H gradient in solution.
l ① lipid soluble diffusion (simple diffusion) l The nonionized molecules are usually lipid soluble and can diffuse across the cell membrane. l ② Filtration through pores l Hydrophilic lipid-insoluble substances can cross membranes through water-filled pores. l ③ Passive facilitated diffusion l This flows the concentration gradient but dose not obey simple diffusion lows. It is believed to involve a carrier mechanism.
l (2) Active transport l This is a process in which a solute moves across membrane against an electrochemical gradient. Either against a concentration gradient or, if the solution is charged. l ※ It transports against a concentration gradient. l ※ It needs metabolic energy l ※ It involves carrier mechanism, so it can become saturated l ※ It shows specificity for a particular type of chemical structure
l 3. Ionicity influence lipid-soluble diffusion l the degree of dissociation of the drugs can express as Handerson -Hasselbalch equation:
weak acids: HA weak bases: H+ + A - BH Ka = [ H + ] [A- ] H+ + B Ka = [ H +] [ B- ] [ HA ] [BH] -lg. Ka=-lg[H+]-lg [A-] [HA] -lg. Ka=-lg[H+]-lg [B] [BH+] pka = p. H - lg [A-] [ HA ] [B] [ BH+ ] That:p. H - pka = lg [ A- ] [ HA ] pka - p. H = lg [ BH ] [B]
10 p. H -pka = [ A- ] [ HA ] When p. H = pka, 10 pka -p. H = [ HA ] [B] when p. H = pka, [ HA ] = [ A ] [ B ] = [ BH ] 10 p. H -pka = [ A- ] 10 pka -p. H = [ HA ] [B] When p. H = pka, when p. H = pka, [ HA ] = [ A ] [ B ] = [ BH ]
3. Process of drugs in the body l (1) absorption l ①absorption of drugs from alimentary canal l oral ingestion is the route of most drugs administration. l Absorption from mouth: gastrointestinal tract epithelium in blood l portal circulation liver systemic circulation
first pass elimination: drug concentration in the blood is declined because metabolism of drugs as a result of passage through the liver and intestine when absorption in the alimentary tract. l rectal administration: 50%of the drainage of the rectal region bypasses the portal circulation; thus, the biotransformation of drugs by the liver is minimized. It is also useful if the drug induces vomiting, when given orally or the patient is already vomiting. l
l ② sublingual administration l Placement under the tongue allows the drug to diffuse into the capillary network and therefore to enter the systemic circulation directly. l Both the sublingual and the rectal rout have the additional advantage that they prevent the destruction of the drug by intestine and liver enzyme or by low p. H in the stomach. l parenteral adminstration
l intravenous injection l subcutaneous injection l intramuscular injection l intra-arterial injection l ① pulmonary absorption l ② transdermal administration l (2) distribution of drugs l Factors influence distribution of drugs
redistribution Blood vessel brain heart liver pseudoequilibrium fat
blood flow and affinity of drugs with tissues l ② physicochemical characteristics l ③ protein binding ※ the binding is reversible and there is a dynamic equilibrium between the bound and unbound forms of a drug. l ①regional
l D D + P DP l ※ Bound drug is not effective and serve as a substantial drug reservoir l ※ The drugs with similar physicochemical characteristics compete with each other for the biding site and result competitive inhibition.
① special barrier l ※ blood-brain barrier l ※ placenta barrier 3. biotransformation of drugs l (1) oxidative reaction l (2) reductive reaction l (3) hydrolytic reaction l (4) counjugation reaction
the hepatic microsomal enzymes: l It is in the endoplasmic reticulum therefore often are classified as microsomal enzymes. These are primarily cytochrome P 450 enzyme family that is the major catalyst of drug biotransformation reactions.
Non-specificity l ※ The activity has some limit, it can be inhibited or induced competitively by other drugs interacting with the same system l ※ It has large difference in various individual l ※ It can be inducted or inhibited by various drugs l The hepatic microsomal enzymes inducers:
The hepatic microsomal enzymes inducers: l The drugs that can increase synthesis and activity of the hepatic microsomal enzymes. It leads to an increased rate of biotransformation and corresponding decreases in the availability of the parent drug. The hepatic microsomal enzymes inhibitors: l The drugs that can decrease synthesis and activity of the hepatic microsomal enzymes.
l Inhibition of drug biotransformation enzymes results in elevated levels of the parent drug, prolonged pharmacological effects, and an increased incidence of drug-induced toxicity. l Competition between two or more drugs for the active site of the same enzyme may lead to a decrease in the metabolism of one of these agents, depending on the relative concentrations of each substrate and their affinities for the enzyme.
4 Excretion of drugs q drugs are eliminated from the body either unchanged or as metablites. l q Excretory organs, the lung excluded, eliminate polar compounds more efficiently than substances with high lipid solubility. q The kidney is the most important organ for elimination of drugs and their metabolite.
(1) Renal excretion l Excretion of drugs and metabolites in the urine involves three processes: glomerular filtration, active tubular secretion, and passive tubular reabsorption. (2) Biliary and fecal excretion l Hepato-enteral circulation: the metabolites of drugs formed in the liver are excreted into the intestinal tract in the bile and are reabsorbed from the intestine.
(3) Excretion by other routes l The metabolites of drugs are also excreted into sweat, saliva, tears, and breast milk. l Although excretion into hair and skin also is quantitatively unimportant, sensitive methods of detection of toxic metals in these tissues have forensic significance.
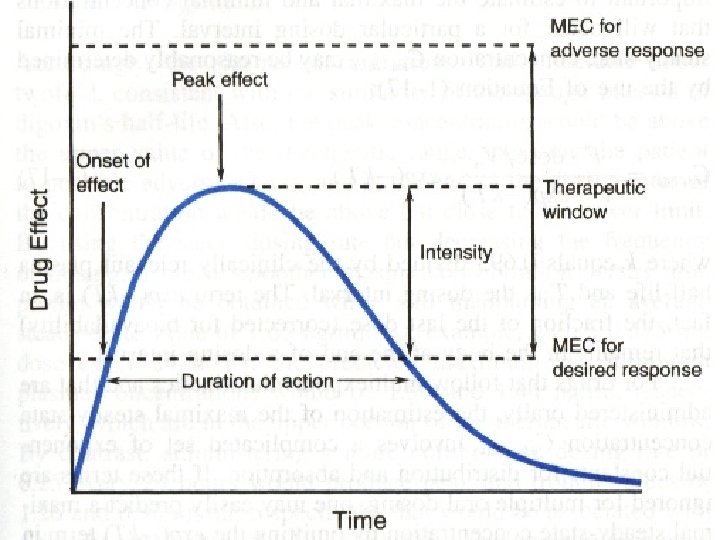
Section 2. time process of drug concentration change in the body 1. time–concentration relationship and timeresponse relationship l Drug concentration time curve (C-T curve): the curve is obtained from concentration or logarithmic concentration for the ordinate and from time for the abscissa.
(1) drug absorption and C-T curve l ① peak concentration (Cmax) l ② peak time (Tmax) l ③ effective period l ④ area under the curve (AUC)
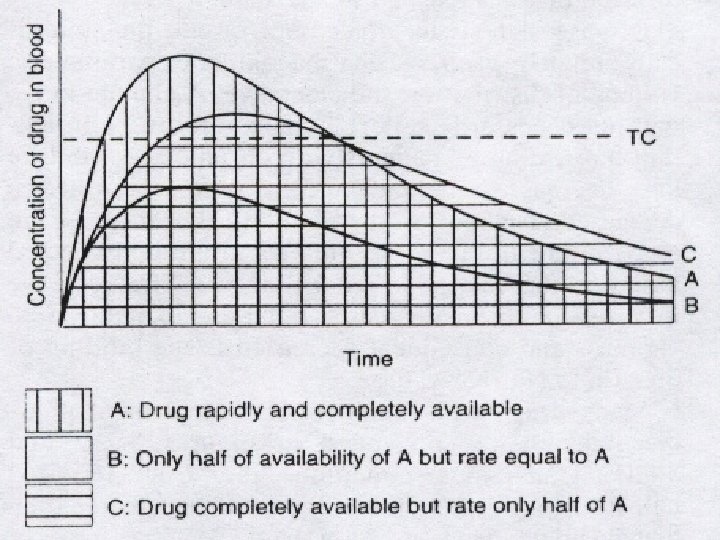
l ⑤ bioavailability l The amount of the drug that reaches the systemic circulation can be expressed as a fraction of the dose F, which often is called bioavailability. AUC(parenteral administration) l F= × 100% AUC(intravenous administration) AUC(trial drug) l F= × 100% AUC(standard drug) l it is important to distinguish between the rate and extent of drug absorption and the amount that ultimately reaches the systemic circulation.
l (1) distribution of the drug and C-T curve l ① one-compartment model l intravenous administration drug Central compartment l l elimination Figure. plasma concentration-time curves following intravenous administration of a drug(500 mg) to a 70 kg man.
l ② two-compartment model intravenous administration drug elimination central compartment l k 21 k 12 distribution l l external compartment l It indicates that, in fact, the drug follows multi- exponential kinetics.
① volume of distribution (Vd) l the volume of distribution (V) relates the amount of drug in the body to the concentration of drug (C) in the blood or plasma, depending upon the fluid measured l This volume dose not necessarily refer to an identifiable physiological volume, but merely to the fluid volume that would be required to contain all of the drug in the body at the same concentration as in the blood or plasma.
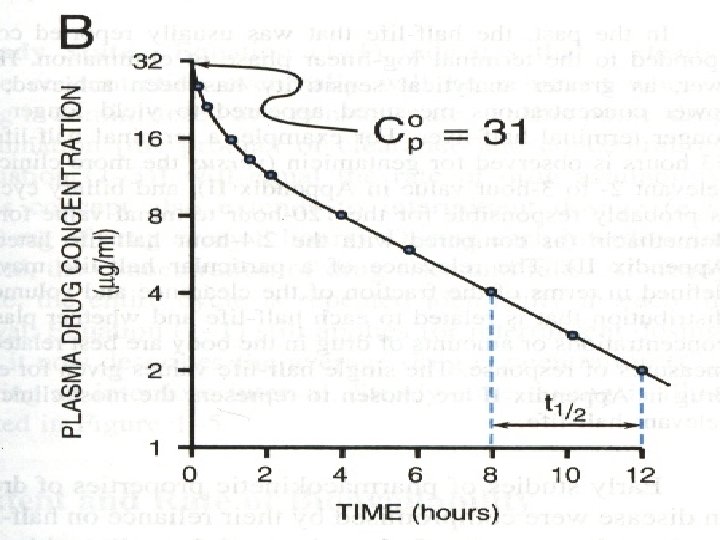
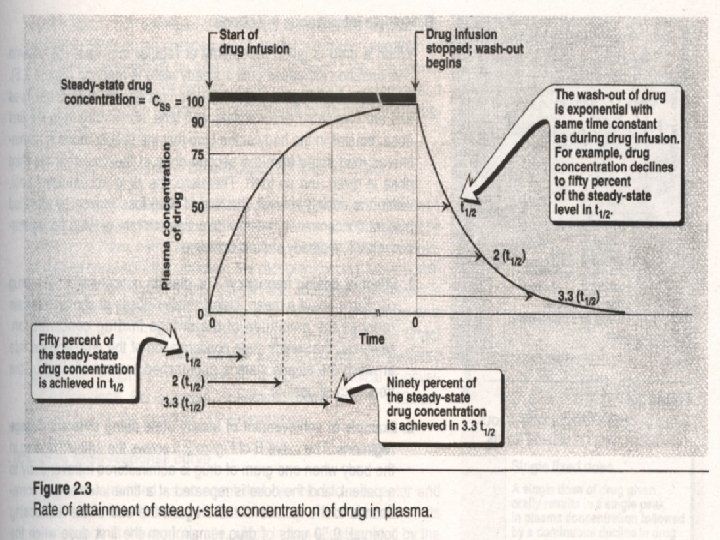
FD Vd = F: bioavailability, D: drug dose, C l C: plasma concentration of drug l l l l Section 3 eliminated kinetics of drug 1. elimination of drug and C-T curve ① half-life (T 1/2): the time it takes for the plasma concentration or the amount of drug in the body to be reduced by 50%. According to first-order kinetics: C = C 0 e-ket, C/C 0=e-ket
l takes the nature logarithm: l Ln(C/C 0)=-ket, t=ln(C/C 0)/-k l when t = 1/2, l t 1/2=ln 1/2/-k=2. 303/-k=0. 693/k l ∴ t 1/2= 0. 693/k l The elimination in various half life is: l At = A 0 e-kt = A 0 e-0. 693/t 1/2×n×t 1/2 = A 0 e-0. 693 n =A 0 (0. 5)n = A 0 (1/2)n l When n = 5, At = 3%A 0 l it shows that drug is almost eliminated from the body.
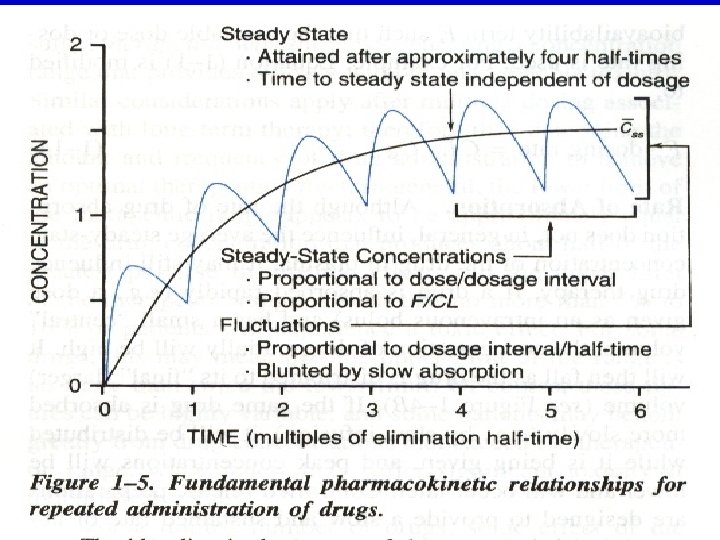
l if the drug repeated administration at intervals equal to its elimination half life time, the steady state concentration (Css) is attained after approximately 5 half life times. l The calculated equation of accumulation volume in various half life is: l At = A 0(1 -e-kt) = A 0(1 -e 0. 693/t 1/2×n) l = A 0(1 -e-0. 693 n) = A 0[1 -(0. 5)n] = A 0[1 -(1/2)n]
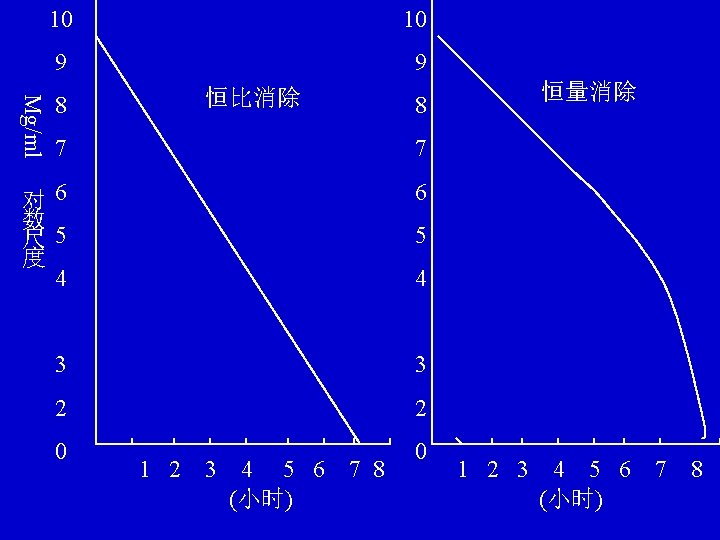
2. first -order kinetic elimination l (1) drug is cleared in an exponential manner and eliminated in a constant fraction; l (2) T 1/2 is constant, l T 1/2 = 0. 693/K. l the steady state concentration (Css) or almost elimination of the drug in the body is attained after approximately 5 half life times.
3. zero -order kinetic elimination l drug is eliminated in a constant volume manner l T 1/2 is not constant, T 1/2 = 0. 5/C 0 l according to the equation: l dc/dt=-k. C 0, l C = C 0 – kt C 0 -C C 0 -1/2 C 0 l T = , t = 0. 5 C 0/k 1/2 = k k l ∴T 1/2 = 0. 5 C 0/k
1. 0 10 0. 9 9 0. 8 8 恒量消除 浓 0. 7 度 0. 6 7 (Mg/ml ) 0. 5 5 0. 4 4 0. 3 3 0. 2 2 0. 1 1 0 0 恒比消除 6 1 2 3 4 5 6 7 8 1 2 3 4 5 6 7
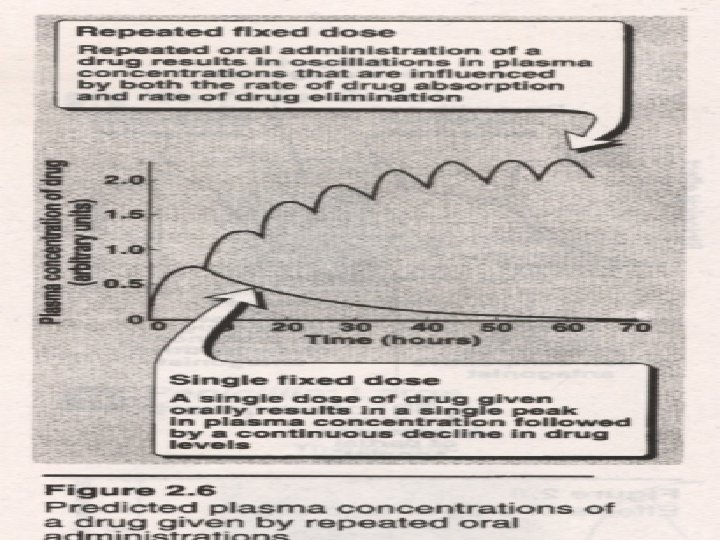
2. repetitive doses and C-T curve l commonly used administration scheme l (1) Repetitive administration in equal doses and at regular interval l (2) administration scheme of loading dose and maintenance doses
① Loading dose l A “loading dose” is one or series of doses that may be given at the onset of therapy with the aim of achieving the target concentration rapidly. l The appropriate magnitude for the loading dose is:
l l l l Dl = Dm/(1 -e-kt) if: t = t 1/2, then: Dl =Dm/1 -e-0. 693 t 1/2÷t 1/2 =Dm/1 -e 0. 693 = Dm/0. 5= 2 Dm A loading dose is chosen which is twice the maintenance dose at first and the following maintenance dose interval is equal to T 1/2 of the drug, the steady state blood concentration (Css) should be established from the beginning. When the interval time of administration τ= T 1/2, Css = (F×dose)÷(Cl×T)
l In most clinical situations, drugs are administered in a series of repetitive doses or as a continuous infusion in order to maintain a steady state concentration of drug in plasma within a given therapeutic range. l To maintain the chosen steady state or target concentration, the rate of drug administration is adjusted such that the rate of input equals the rate of loss. l Dm = (MTC – MEC)×Vd
l loading dose administration scheme of intravenous progression dripping injection in even rate: l Css =R(1 -e-ket)/CL l When beginning: t = 0, e-ket =1, (1 -eket) = 0, l when arrives in Css, e-ket → 0, than (1 -eket)→ 1, C =R/CL ss l from CL = Vd� k to Css =R/CL: l Css =R/Vd� k l ∵ k =0. 693/k , or 1/k=1. 44� t 1/2 , l ∴ Css = R/Vd� t 1/2/0. 693 =R/Vd� 1. 44