Universidade Federal de Gois UFG AULA DE FARMACOLOGIA



 Fase Farmacêutica Fonte: http: //www. fotosearch. com/u 13272187 DROGA Fase")






")

")

 Dupla camada (lipídeos) com cadeias de hidrocarboneto orientadas para dentro,")
")
 Drogas Ligadas não Atravessam as membranas Drogas Lipofílicas Acumulam-se")


 Mucosa intestinal")
 EQUAÇÃO DE HENDERSON-HASSELBALCH ácido base “mais ionizados,")







 para o extravascular (intersticial). • Do plasmático para")






 e")



 também")





: É o volume no qual a droga")

")










 apresentam taxa de eliminação intermediária entre as")













 é conjugado com a droga")












 TUBO CONTORCIDO DISTAL GLOMÉRULO")






















 Cl. T")




- Slides: 114
Universidade Federal de Goiás – UFG AULA DE FARMACOLOGIA Farmacocinética Prof. MS. Farm. Hugo Campos Oliveira Santos Goiânia, Fevereiro de 2013.
FARMACOLOGIA Conceito & Definição Pharmakon + Logos É o estudo das substâncias que interagem com sistemas vivos por meio de processos Químicos. Fonte: www. istockphoto. com Farmacologia engloba o conhecimento da história, origem, propriedades físico-químicas, associações, efeitos bioquímicos e fisiológicos relacionado aos medicamentos. Fonte: Katzung, 2003.
FARMACOLOGIA DEFINIÇÕES Droga : substância química capaz de modificar a função dos organismos vivos, com finalidades medicamentosa ou sanitária. Fármaco: designa uma substância química conhecida e de estrutura química definida dotada de propriedade farmacológica. Medicamento: produto farmacêutico, tecnicamente obtido ou elaborado, com finalidade profilática, curativa, paliativa ou para fins de diagnóstico. Fonte: Lei nº 5. 991, de 17 de dezembro de 1973. Fonte: http: //www. betadaily. com
FARMACOLOGIA FASES FARMACOLÓGICAS (RESUMO) Fase Farmacêutica Fonte: http: //www. fotosearch. com/u 13272187 DROGA Fase Farmacocinética Vias de Administração Absorção Distribuição Biotransformação Eliminação ORGANISMO Fase Farmacodinâmica Efeitos Fisiológicos Efeitos Bioquímicos Mecanismo de Ação Concentração no local do receptor
FASE FARMACÊUTICA Investigação Farmacológica Fase pré-clínica Estudos em animais Fase clínica Fase I Voluntários sadios (Humanos) (Dose, Avaliação, Efeitos Adversos) Fase II Dosagem mais segura e eficaz Fase III Triagem clínica Eficácia terapêutica e segurança Indicação Clínica / Regulamentação Vigilância Pós-comercialização Fonte: WWW. GOOGLE. COM
CONCEITOS BÁSICOS Biodisponibilidade Corresponde à fração do fármaco administrada que alcança a circulação sistêmica, incluindo a sua curva de concentração e tempo na circulação sistêmica. Medicamento similar – aquele que contém os mesmos princípios ativos, apresenta a mesma concentração, forma farmacêutica, via de administração, posologia e indicação. Identificado: nome comercial ou marca. Medicamento inovador – medicamento apresentando em sua composição ao menos um fármaco ativo que tenha sido objeto de patente. Medicamento de referência – medicamento inovador registrado (ANVISA) cuja eficácia, segurança e qualidade foram comprovados cientificamente. Medicamento genérico (intercambiável) geralmente produzido após a expiração ou renuncia da proteção patentária ou de outros direitos de exclusividade, comprovada a sua eficácia, segurança e qualidade. Designado pela (DCB) ou, na sua ausência, pela (DCI). Resolução n 0 391 de 09/08/99
FASE FARMACOCINÉTICA Definição Área da Farmacologia que estuda os processos de absorção, distribuição, biotransformação e excreção dos fármacos, ou seja, o que o organismo faz sobre o fármaco. Considera o caminho que o medicamento faz no organismo Movimento do fármaco “in vivo” Fonte: http: //images. veer. com
FARMACOCINÉTICA CLÍNICA Ajuste da terapêutica em diferentes pacientes, tanto da resposta desejada quanto da toxicidade em função da concentração do fármaco em seu sítio de ação. Produto Farmacêutico Industria Farmacêutica Liberação (Desintegração) Dissolução (Dispersão Molecular) Lei n 0 9787/99 ANVISA Biodisponibilidade Interação Fármaco / Organismo Genéricos Concentração Plasmática Fonte: http: //www. fotosearch. com Forma farmacêutica
Fonte: http: //2. bp. blogspot. com/_r. T 2 z. KLK 32 o. U/TBVwgv. Mga 6 I/AAAAAWI/Nwm. F 3 We-g. M 0/s 1600/medicamento. jpg CONCENTRAÇÃO TERAPÊUTICA Concentrações de efeito mínimo eficaz (limite mínimo) e efeito tóxico (concentração máxima tolerada, limite máximo). “Janela Terapêutica" Faixa de Concentração Eficácia/Toxicidade INSUFICIENTE Sítio de Ação - Receptores Dose & Intervalo de Administração ADEQUADO I. T. EXCESSO Amitriptilina – Faixa Estreita (I. T. < 5) IT: Índice Terapêutico Descartado erroneamente Efeito Desejável [ ] terapêutica Toxicidade
FARMACOCINÉTICA VIAS DE ADMINISTRAÇÃO VIA ORAL Vantagem: Seguro/Econômico Desvantagem: Patologias do sistema digestivo, p. H gástrico e presença de alimentos/absorção Fonte: http: //www. ff. up. pt/toxicologia/monografias
OUTRAS VIAS DE ADMINISTRAÇÃO Sublingual: Absorção através de pequenos vasos sangüíneos. Exemplo: Nitroglicerina (Angina) Retal: Uso em quadros de vômitos, tratamento local ou inconscientes. Evita 1ª Passagem (Fígado): Fármacos (Veia Cava Inferior) Supositórios: absorção irregular e irritação local. IV – Intravenosa: não pode ser: hemolítica, cáustica, não coagular as albuminas, não produzir embolia ou trombose, não conter pirogênio. Aplicação: Deve ser administrada lentamente e com monitorização constante IM - Intramuscular Tópica – (mucosa, pele e olho) Intratecal Absorção Pulmonar Intra-arterial Intraperitoneal
VIAS DE ADMINISTRAÇÃO Concentração - Pico
PROCESSOS FARMACOCINÉTICOS (Resumo)
ABSORÇÃO Passagem da droga do seu local de aplicação até a corrente sangüínea. Fonte: http: //www. fotosearch. com/u 13132187 Fatores Envolvidos da Absorção: Ligados aos Medicamentos Concentração Lipossolubilidade Peso Molecular Grau de Ionização Via de Administração, características fisiopatológicas, idade, sexo, peso corporal e raça do paciente. Ligados ao Organismo Vascularização Superfície de Absorção Permeabilidade Capilar
Membranas Celulares (Revisão) Dupla camada (lipídeos) com cadeias de hidrocarboneto orientadas para dentro, (característica hidrofóbica). As proteínas exercem função de receptores que proporcionam vias de sinalização elétricas ou químicas e alvos seletivos para a ação de fármacos.
Mecanismos de transporte através de membranas Transporte passivo Transporte ativo Transporte facilitado Pinocitose (líquidos) Fagocitose (sólidos)
Transporte através de Membranas (Fármacos) Drogas Ligadas não Atravessam as membranas Drogas Lipofílicas Acumulam-se Tecido Adiposo “Mal Nutrição” Albumina + Droga livre
Utilizadas por Moléculas Não-Polares Ex: Anestésicos, tranquilizantes, Antibióticos, hormônios e sedativos. DIFUSÃO Difusão Simples Difusão Facilitada: Participação de molécula transportadora (permeases), não há consumo de energia , substância move-se de acordo com o gradiente de concentração Transporte Passivo/filtração (Lipossolubilidade) Difusão Facilitada Fonte: http: //farm 3. static. flickr. com
Transporte ativo: células do túbulo renal, trato biliar, barreira hematoencefálica e TGI. Transporte Ativo Fonte: http: //djalmasantos. files. wordpress. com/2010/09/ativo. jpg Há gasto de ATP (adenosina trifosfato) perdendo um fosfato e virando ADP (adenosina difosfato) – Contra o Gradiente de Concentração (Bomba Na+/K+)
p. H dos compartimentos biológicos Mucosa gástrica – p. H 1 (aproximadamente) Mucosa intestinal – p. H 5 Plasma – p. H 7, 4 A equação de Henderson-Hasselbach pode ser empregada na previsão do comportamento farmacocinético de fármacos HA H 3 O + + A Meio extracelular Meio intracelular
“Constante “ pka “grau de dissociação” (ionização) EQUAÇÃO DE HENDERSON-HASSELBALCH ácido base “mais ionizados, excreção fácil “ “pouco ionizada” Fármacos com caráter ácido se acumulam no compartimento com p. H mais básico e fármacos com caráter básico se acumulam no compartimento com p. H mais ácido. Ácido fraco, terá boa absorção num meio ácido (p. H ácido), pois, há uma ionização menor, AAS.
Relação p. H e pka Polaridade Molecular, ionização e ph do meio: ph > pka Ácido: Forma ionizada Base: Forma não- ionizada (B) ph < pka Ácido: não – ionizado (HA) Base: ionizado
Pka – Bases e Ácidos Fonte: http: //www. sistemanervoso. com/images/farma/fac_06. gif
Ácido Acetil Salicílico - AAS Ácido orgânico fraco, p. Ka 3, 7 HASac + H 2 O = ASac- + H 3 O+ Rapidamente absorvido no estômago. Eliminado na forma de saliciliato. Alcalinização da urina aumenta a eliminação de salicilato. Inibe a biossíntese das prostaglandinas !
Anestésicos Locais Base Fraca - p. Ka em torno de 8 a 9 Bloqueiam de modo reversível a condução de impulsos ao longo dos axônios dos nervos e outras membranas excitáveis que utilizam canais de sódio com principal meio de geração de potenciais de ação. Xilocaína
LIPOFILIA E LATÊNCIA Coeficiente de partição: Coloca-se o fármaco num meio com 2 fases: aquosa e lipofílica; agita-se e mede-se a quantidade dissolvida. > Lipofilia < latência Potência: Capacidade da molécula interferir na estrutura e de inibir o funcionamento dos canais iônicos. Relacionada com a ligação (proteínas e lipofilia) de um determinado composto. Quanto maior for a ligação das proteínas plasmáticas menor será a potência e menor a sua toxicidade Quanto maior a fração não ionizada, maior é a facilidade de penetração => maior potência (menor latência). Quanto maior for a lipofilia (coeficiente de partição), maior será a potência (melhor penetração). Latência (tempo decorrido entre administração e efeito).
PRÓ-FÁRMACO Introdução de novos fármacos na terapêutica através dos processos de modificação molecular. Latenciação: transformação do fármaco em forma de transporte inativo que, “in vivo”, mediante reação química ou enzimática, libera a porção ativa no local de ação. Uso: quimioterápicos específicos contra os maiores desafios da Ciência na atualidade: AIDS e câncer Fonte: CHIN & FERREIRA. 1999.
INFLUÊNCIA DO PH NA ABSORÇÃO E DISTRIBUIÇÃO DE DROGAS: pka p. H • drogas ácidas: ionizam-se pouco no estômago, portanto são bem absorvidas. Ionizam-se quase completamente no intestino ou no sangue, permanecendo nesses compartimentos. • drogas básicas: ionizam-se quase completamente no estômago, não sendo bem absorvidas pela mucosa estomacal. Ionizam-se muito pouco no intestino ou no sangue, sendo absorvidos pela mucosa intestinal.
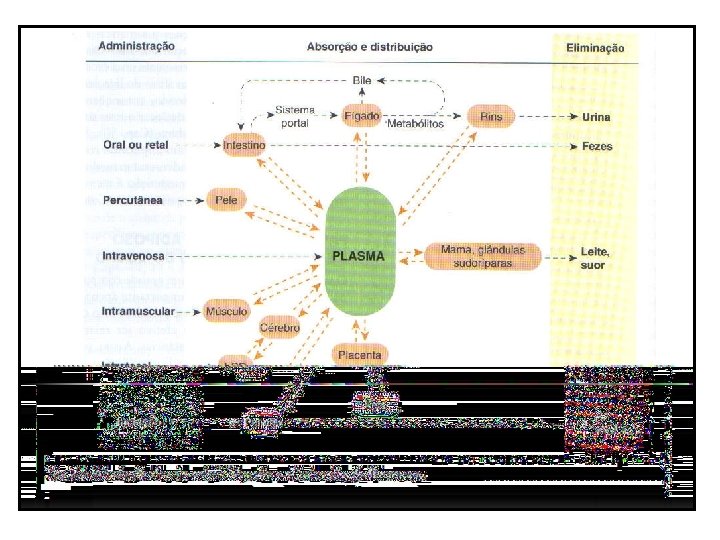
DISTRIBUIÇÃO • Do compartimento intravascular (plasmático) para o extravascular (intersticial). • Do plasmático para o cefalorraquidiano. • Do plasmático para o extravascular e intracelular cerebrais. • Do plasmático para o placentário. • Do plasmático para o tubular renal. • Do plasmático para o intracelular.
Distribuição Pediátrica • Composição Corporal – água total & fluido extracellular – tecido adiposo & músculo esquelético • Ligação às Proteínas – albumina, bilirrubina, 1 -ácido glicoproteína • Ligação aos Tecidos – Variações da composição
DISTRIBUIÇÃO • a droga que atinge a corrente sanguínea será distribuída a diferentes partes do organismo, em uma taxa que depende de vários fatores.
VELOCIDADE DE FLUXO SANGUÍNEO: • Determina a quantidade máxima de droga que pode ser distribuída por minuto para órgãos e tecidos específicos, para uma dada concentração plasmática. • Tecidos mais ou menos irrigados receberão diferentes quantidades da droga; também o tempo de permanência das drogas nos tecidos irá variar.
FATORES ANATÔMICOS E FISIOLÓGICOS: • Inicialmente, fígado, rins, cérebro e outros órgãos com boa perfusão recebem a maior parte do fármaco. • A liberação para músculos, a maior parte das vísceras, pele e gordura a liberação é mais lenta. • Essa 2ª fase de distribuição pode levar minutos ou várias horas antes que a concentração do fármaco nos tecido esteja em equilíbrio com o sangue.
FATORES ANATÔMICOS E FISIOLÓGICOS: • A segunda fase também envolve uma parte maior de massa corporal que a inicial e geralmente responde pela maior parte do fármaco distribuído. • A lipossosubilidade é um importante fator para a difusão do fármaco. • O p. H gradiente de p. H entre líquido intra e extracelular (7, 0 versus 7, 4). • O principal fator é a ligação do fármaco com proteínas plasmáticas.
FATORES ANATÔMICOS E FISIOLÓGICOS: • Barreira hematoencefálica → ausência de fenestrações e presença de tight junctions nos capilares cerebrais. Dificulta a passagem principalmente de substâncias hidrossolúveis, ionizadas ou polares. Glicose, aminoácidos, aminas, purinas e ácidos orgânicos → transporte ativo. • A menos que contrariamente comprovado, devese pressupor que todas as drogas atravessam a placenta e também penetram nas glândulas mamárias.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • As principais proteínas são: Albumina (ácidos) e α 1 - glicoproteína ácida (bases). Fenilbutazona se liga 95%. • O grau de ligação proteica das drogas depende de: afinidade entre droga e proteínas; concentração sanguínea da droga; concentração sanguínea das proteínas.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • Contribui para as diferenças nas concentrações das drogas em diferentes compartimentos corporais. • A ligação à proteínas impede a ação do fármaco e geralmente limita o transporte e o metabolismo do mesmo. No entanto, a ligação plasmática geralmente não limita a secreção tubular renal e a biotransformação.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • Em baixas concentrações de fármaco, a parte ligada é proporcional à concentração nos locais de ligação e à constante de dissociação. • Em altas concentrações de fármaco, a parte ligada é proporcional ao número de locais de ligação e à concentração do fármaco.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • Portanto, a ligação plasmática é um processo saturável e não-linear. • No entanto, para a maioria dos fármacos, a faixa terapêutica de concentração plasmática é limitada, de modo que a extensão das partes ligadas e livres é relativamente constante.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • A extensão da ligação plasmática (proteínas) também podem ser alterada por fatores relacionados com a doença. Por exemplo: a hipoalbuminemia secundária a doença hepática grave ou a síndrome nefrótica levam à diminuição da ligação e o aumento da fração livre.
LIGAÇÃO DAS DROGAS A PROTEÍNAS PLASMÁTICAS: • Do mesmo modo, as afecções que levam a uma resposta de reação de fase aguda (câncer, artrite, infarto do miocárdio) levam a altos níveis de α 1 - glicoproteína ácida e aumento da ligação de fármacos básicos.
LIGAÇÃO TECIDUAL • Muitos fármacos se acumulam nos tecidos em contrações maiores que aquelas dos líquidos extracelulares e do sangue. • Uma grande fração do fármaco pode estar ligado desse modo e servir de reservatório que prolonga a ação do fármaco. • Tecido adiposo (70% do barbitúrico tiopental – 3 hs após a administração) • Ossos (tetraciclinas, substâncias tóxicas como chumbo, rádio)
REDISTRIBUIÇÃO • O fármaco se redistribui para outro tecido, sendo um fator de término da ação do mesmo. • Um bom exemplo disso é o uso intravenoso do fármaco tiopental que alcança sua contração máxima no cérebro em 1 minuto após sua injeção. • Redistribuição em cadáveres.
PERMEABILIDADE CAPILAR • As drogas atravessam as paredes capilares por duas vias: • Via Transcelular, droga atravessa a célula endotelial por pinocitose, por difusão simples ou transporte ativo. • Via intercelular, a travessia é feita poros ou canais existentes no endotélio e entre células.
VOLUMES REAL E APARENTE DE DISTRIBUIÇÃO • VOLUME REAL: Se a droga tiver capacidade de atravessar as membranas celulares o volume real para uma pessoa de 70 Kg é de aproximadamente 43 Litros. Plasma = 3 L Líquido intersticial extravascular = 12 L Líquido intracelular = 28 L
• VOLUME APARENTE DE DISTRIBUIÇÃO (Vd): É o volume no qual a droga teria que se dissolver, a fim de atingir a mesma concentração que ela se encontra no plasma. Nessa definição, a concentração plasmática da droga é aquela observada após a absorção e distribuição e antes da eliminação.
• • Exemplos: Antidepressivos Tricíclicos Vd= 20 L/Kg Varfarina Vd= 0, 1 L/Kg Vd elevados indicam que as drogas possuem grandes concentrações teciduais.
• Fórmula: • Vd= quantidade de fármaco no corpo C (concentração do fármaco) Exemplo: 500µg de digoxina pessoa de 70 Kg Cocntração plasmática de 0, 75 ng/ml Vd= 500 Vd= 667 litros 0, 75
VOLUMES REAL E APARENTE DE DISTRIBUIÇÃO Esses volumes podem variar de indivíduo para indivíduo de acordo com muitos fatores: • Dependentes da droga: - lipossolubilidade - polaridade, ionização - grau de ligação com proteínas plasmáticas ou com proteínas teciduais
VOLUMES REAL E APARENTE DE DISTRIBUIÇÃO • Dependentes do paciente: - idade - peso e tamanho corporal - hemodinâmica - concentração das proteínas plasmáticas - estados patológicos - genética
CONCENTRAÇÃO PLASMÁTICA DAS DROGAS • A constância da concentração plasmática máxima média é contingencial e reflete um estado estável de equilíbrio dinâmico entre a dose da droga que é administrada e a taxa da droga que é distribuída e eliminada.
CONCENTRAÇÃO PLASMÁTICA DAS DROGAS • Tendo-se conhecimento da concentração plasmática indicada pela terapêutica, o ajuste posológico é estabelecido de dois modos: 1 - Com uma dose inicial, de ataque, seguida por uma dose de manutenção. 2 - Com uma série de doses seguidas até que, após quatro a seis meias-vidas da droga, se atinja a concentração sanguínea máxima constante média da droga em questão.
VARIAÇÃO DA CONCENTRAÇÃO PLASMÁTICA DAS DROGAS • As variações individuais são originadas nas biotransformações, na absorção, na distribuição, na excreção, na biodisponibilidade, na patologia renal, hapática, tireoidiana, na cardíaca e na interação com outras drogas.
IMPLICAÇÕES CLÍNICAS DA VARIAÇÃO DA CONCENTRAÇÃO PLASMÁTICA DAS DROGAS • A farmacocinética atual já dispõe de experiências para resolver problemas dos seguintes tipos em que a concentração plasmática da droga desempenha papel essencial: a. determinação da posologia adequada de drogas que possuem meia-vida curta, como a procainamida e alprenolol (t½ = 2 a 3 horas).
IMPLICAÇÕES CLÍNICAS DA VARIAÇÃO DA CONCENTRAÇÃO PLASMÁTICA DAS DROGAS b. ajuste posológico de drogas cujas meias-vidas, como a digoxina (t½ = 30 a 40 horas) e a gentamicina (t½ = 2 a 3 horas), são prolongadas pela insuficiência renal. c. uso de dose de ataque , como se faz, por exemplo, com a digoxina quando há necessidade de efeito rápido, encurtando-se o tempo necessário para alcançar a concentração plasmática constante média; nesse caso, é necessário conhecer o volume aparente de distribuição da droga.
IMPLICAÇÕES CLÍNICAS DA VARIAÇÃO DA CONCENTRAÇÃO PLASMÁTICA DAS DROGAS d. possibilidade de prever a concentração plasmática máxima constante média após doses repetidas. e. a não-obediência, por parte do paciente, ao esquema poslógico. f. biodisponibilidade da droga. g. ajuste posológico de drogas usadas profilaticamente. h. ajuste posológico em pacientes cuja resposta não é compatível com o quadro clínico nem com a dose administrada.
EQUAÇÃO DA CONCENTRAÇÃO PLASMÁTICA DAS DROGAS Css = F. Dose Vd. Kel. T Css = concentração plasmática máxima constante média F = fração da dose que alcança a circulação sistêmica (biodisponibilidade) Kel = constante de eliminação de primeira orcdem Vd = volume aparente de distribuição T = intervalo em horas entre doses
ELIMINAÇÃO – METABOLISMO E EXCREÇÃO: • Cinética de primeira ordem: a velocidade de remoção da droga do organismo é proporcional à concentração plasmática da mesma. A maioria das substâncias obedece a essa cinética. • Cinética de ordem zero: algumas poucas drogas (etanol por ex. ) são eliminadas a uma velocidade constante, não havendo influência da concentração plasmática.
• Outras drogas (aspirina e fenitoína) apresentam taxa de eliminação intermediária entre as cinéticas de ordem zero e primeira ordem. • Pequenas modificações na dose podem levar a aumentos desproporcionais na concentração plasmática resultando em toxicidade.
METABOLISMO DAS DROGAS • O metabolismo é um processo alternativo que pode levar ao término da atividade biológica ou à sua alteração. Em geral os xenobióticos lipofílicos são transformados em substâncias mais polares e, portanto passível de excreção mais fácil. • Drogas e toxinas são agentes estranhos ao organismo.
METABOLISMO DAS DROGAS • Drogas podem ser metabolizadas nos pulmões, sangue e fígado • O organismo converte as drogas em formas menos ativas e aumenta a sua hidrossolubilidade para melhorar a eliminação • Como por exemplo, barbitúricos lipossolúveis como o tiopental e o pentobarbital, teriam meias-vidas extremamente longas, não fosse seu metabolismo.
METABOLISMO DAS DROGAS • Por outro lado, substâncias lipofílicas, como DDT, que são armazenadas no tecido adiposo e protegidas dos principais órgãos envolvidos no metabolismo de drogas, podem persistir na gordurante anos. • Também foram elaboradas pró-drogas farmacologicamente inativas.
METABOLISMO DAS DROGAS • Fígado – principal via do metabolismo das drogas • Fígado pode também converter pró-drogas (inativas) na sua forma ativa • Tipos de reações – Fase I (sistema do Citocromo P 450) – Fase II
Reações de Fase I Reações não-sintéticas ou catabólicas. • Em geral as reações de fase I convertem a droga original num metabólito mais polar, ao induzir ou expor um grupo funcional (-OH, NH 2, -SH). Com freqüência, esses metabólitos são inativos, ou sua atividade é apenas modificada.
Tipos de Reações de Fase I • • • Hidrólise Oxidação Redução Demetilação Metabolismo da Álcool-desidrogenase
Oxidação • Consiste na adição de oxigênio ou de um radical carregado negativamente, ou então na remoção de hidrogênio ou de um radical carregado positivamente. • Nesse processo são utilizadas duas enzimas. A primeira é a NADPH – citocromo P 450 redutase e a segunda é uma hemoproteína chamada citocromo P 450, que funciona como a oxidase terminal.
Redução • É o inverso da oxidação e envolve as enzimas do citocromo P 450, que então agem em direção oposta àquela observada na oxidação. • Exemplo de droga que é reduzida: cloranfenicol.
Hidrólise • Essa reação consiste na clivagem da molécula da droga pela junção da água. • As amidas e polipeptídeos dão hidrolisados por amidases e peptidases. A hidrólise ocorre no fígado, intestino, plasma e outros tecidos • Exemplos de drogas: procaína, lidocaína e oxitocina.
Ciclização • Nesse caso, forma-se uma estrutura cíclica a partir de um composto de cadeia alifática, como observa com o proguanil (proguanil tiazina)
Desciclização • Há uma abertura da estrutura em anel de moléculas cíclicas drogas, como é o caso dos barbitúricos e as fenitoína.
Reações de Fase I • Sistema do Citocromo P 450 • Localizado no retículo endoplasmático dos hepatócitos • Através de cadeia transportadora de electrões, a droga liga-se ao sistema CYP 450 e entra em oxidação ou redução
METABOLISMO DAS DROGAS: • Fase 1: oxidação, redução, hidrólise. • Adição de um grupo funcional que aumenta a polaridade, e consequentemente, a hidrossolubilidade = diminui a absorção;
Reações de Fase II • Grupo Polar (substância endógena) é conjugado com a droga • Resulta no aumento da polaridade da droga • Tipos de Reacções – Conjugação glucoronídio É a reação de fase II mais importante, compostos que possuem grupos hidroxílico ou carboxílico conjugam-se facilmente com o ácido glicurônico, que é um derivado da glicose. Exemplos de drogas que sofrem essa reação: clorafenicol, Aspirina, fenacetina, morfina metronidazol
– Conjugação da glicina O reagente endógeno é a glicina Ex: ácido salicílico – Conjugação com glutation Nessa conjugação forma-se um mercapturato. Essa reação serve para inativar substâncias altamente reativas, como a quinona e derivados do paracetamol.
– Conjugação do sulfato Pode provocar a inativação de certos compostos como o minoxidil. – Acetilação Os compostos que possuem radicais amínicos e hidrazídicos são conjugados com auxílio da acetil coenzima – A. Ex: sulfonamidas, isoniazida e a hidralazina. – Metilação As enzimas e os fenóis podem ser metilados, como, por exemplo, adrenalina, histamina e ácido nicotínico.
METABOLISMO DAS DROGAS: • Fase 2: conjugação ou síntese. • Adição de um grupamento grande à molécula = facilita a excreção do metabólito.
Genética e Metabolismo das Drogas • Diferenças individuais na expressão gênica das enzimas hepáticas resultam em diferenças individuais na resposta a um medicamento. – Ausência de enzimas – resposta exacerbada à droga; – Excesso de enzimas – resistência inata. Outros fatores podem interferir no metabolismo das drogas: q Dieta e fatores ambientais q. Idade e sexo
Indutores enzimáticos: • A síntese enzimática elevada como resultado da presença de um composto exógeno é chamado de indução. • Substâncias (inclusive algumas drogas e alimentos) que aumentam o metabolismo de outras drogas. • Drogas que promovem indução do seu próprio metabolismo → alto potencial de desenvolvimento de dependência (ex. : barbitúricos, nicotina, etc. ).
Inibidores enzimáticos: • Quando duas drogas competem pela mesma enzima para o metabolismo para uma ou ambas as drogas, chamamos de inibição. • Um exemplo clinicamente importante dessa situação são as arritmias cardíacas, ou convulsões, produzidas pela teofilina quando essa droga é ministrada concomitantemente com um antibiótico macrolídeo como a eritromicina.
• Indutores Etanol, omeprazol, fenibarbital, rifampicina, tabagismo • Inibidores Cimetidina, eritromicina, suco de pomelo, cetoconazol, quinidina
• Interações de drogas Muitos substratos, em virtude de sua lipofilicidade relativamente alta, são retidos não apenas no local ativo da enzima, como também permanecem ligados inespecificamente à membrana lipídica do retículo endoplasmático. Neste estado, podem induzir enzimas microssomais; dependendo dos níveis residuais da droga no local ativo, podem também inibir competitivamente o metabolismo de uma droga administrada simultaneamente.
• O fígado é o principal local de metabolismo das drogas – metabolismo de 1ª. Passagem.
EXCREÇÃO: • CLEARENCE Em geral define-se o clearence sanguíneo ou plasmático de um medicamento por um órgão como o volume sanguíneo ou plasmático totalmente livre da substância por unidade de tempo.
EXCREÇÃO: • CLEARENCE TOTAL Clearence renal + Clearence extra-renal
EXCREÇÃO: • Urina e fezes são as vias mais comuns de excreção; • Bile, pulmões, leite e suor também são vias de excreção.
MECANISMO TUBO CONTORCIDO PROXIMAL- secreção ativa (medicamento livre e ligado) TUBO CONTORCIDO DISTAL GLOMÉRULO FILTRAÇÃO GLOMERULAR ALÇA DE HENLE CANAL COLETOR (medicamento livre) Reabsorção passiva (medicamento lipossolúvel não ionizado)
MECANISMO A figura anterior dá uma representação da estrutura funcional do rim: o néfron. A formação da urina se faz pela atuação de três mecanismos: • filtração glomerular ao nível do glomérulo, não permitindo a difusão de compostos de baixo peso molecular, excluindo as proteínas, principalmente a albumina.
MECANISMO • Secreção tubular permitindo a passagem direta de substâncias de células em direção ao tubo onde se forma a urina. • Reabsorção tubular que permite às substâncias que tenham sido filtradas, passar novamente pelas células e desaparecer da urina. Então um medicamento pode ser filtrado, secretado ou reabsorvido.
Excreção renal: • Filtração glomerular e secreção tubular ativa. A excreção renal de metabólitos está diretamente relacionada à capacidade de excreção de creatinina. • Alterações no p. H ou no fluxo urinário interferem na excreção renal das drogas.
• Variação do p. H pode aumentar, por exemplo, a fração da droga nãoionizada, promovendo reabsorção tubular. • O aumento do fluxo diminui o tempo de contato, diminuindo a reabsorção tubular.
Influência do p. H Urinário • Para os ácidos: ►A influência a influência do p. H urinário é nula devido a substâncias de p. Ka igual ou inferior a 2; com efeito, encontram-se em sua totalidade na forma ionizada seja qual for o p. H e nunca são reabsorvidas.
Influência do p. H Urinário ► Os compostos cujo p. Ka é superior a 8 (ácidos fracos) existem essencialmente na forma não ionizada para os valores de p. H urinário e sofrem um processo de reabsorção intenso e permanente e induzido a um clearence renal fraco. Obs: o p. H urinário é entre 4, 5 e 7, 5.
Influência do p. H Urinário ► A influência do p. H surge como a mais importante para substâncias cujo valor de pka situa-se entre 3, 0 e 7, 5; o clearence renal depende, portanto, do p. H devido a variações da reabsorção. • Para Bases: ► Os compostos com p. Ka superior a 12, 0 (bases fortes) existem sempre na forma ionizada seja qual for o valor do p. H; não há ou há pouca reabsorção.
Influência do p. H Urinário ► As bases mais fracas (p. Ka < 6, 0) e não polares encontram-se sobretudo sob a forma não ionizada para valores de p. H urinário e são constantemente reabsorvidas; seu clerarence renal é fraco. ► As bases fracas polares nunca são reabsorvidas.
Influência do p. H Urinário ► A influência do p. H é determinante para os compostos de p. Ka compreendido entre 6, 0 e 12, 0; a reabsorção varia de um valor nulo a um algarismo muito elevado, de acordo com o p. H. Enfim, os compostos cuja reabsorção varia com o p. H são igualmente sensíveis às modificações do fluxo urinário.
Excreção pelo trato digestivo: • Tempo de trânsito intestinal. • Alguns metabólitos de drogas excretados na bile são hidrolisados, liberando a droga original, resultando em reabsorção e prolongamento do efeito → circulação enteroepática.
Excreção pelo trato digestivo: Características do composto: • A estrutura química • A polaridade • A massa molar Obs: a eliminação biliar é diretamente proporcional à polaridade de uma substância e contém funções ionizáveis. Para ratos é possível estabelecer uma massa molar que abaixo dela a eliminação é impossível.
Excreção pelo trato digestivo: • É grande a semelhança entre este mecanismo e aquele relacionado à secreção urinária dos medicamentos. Ele: - Necessita de um aporte energético. - Pode estar saturado - Pode induzir a fenômenos de competição.
Excreção pelos pulmões: • Via principal de eliminação dos anestésicos gerais. • O etanol também, mas não preferencialmente (teste do “bafômetro”).
Eliminação Pediátrica • Filtração Glomerular amadurece com a idade, valores dos adultos atingidos pelos 3 anos de idade • Recém-nascido = fluxo sanguíneo renal, filtração glomerular e função tubular diminuídas, o que atrasa a eliminação das drogas • Aminoglicosidos, cefalosporinas, penicilinas = intervalo entre doses superior
Princípios Farmacocinéticos • “Steady State”: a quantidade de droga administrada é igual à quantidade de droga eliminada dentro de um intervalo de doses, resultando num plateau ou nivel sérico da droga constante • Drogas com semi-vida curta atingem o “steady state” rapidamente; drogas com semi-vida longa demoram dias a semanas a atingir o “steady state”
Farmacocinética “Steady State” Semi-Vida = tempo necessário para as concentrações no plasma diminuírem em metade (50%) 4 -5 semi-vidas para atingir o “steady state”
Farmacocinética Linear • Linear = a velocidade de eliminação é proporcional à quantidade de droga presente • Aumento da dose resulta num aumento proporcional dos níveis plasmáticos da droga
Farmacocinética Não Linear • Não linear = a velocidade de eliminação é constante independentemente da quantidade de droga presente • Aumentos de dosagem aumentam a saturação dos locais de ligação e resulta em aumento/diminuição nãoproporcionais dos níveis da droga
Cinética Michaelis-Menten • Segue uma cinética linear até as enzimas estarem saturadas • Enzimas responsáveis pelo metabolismo /eliminação tornam-se saturadas resultando em aumento não proporcional dos níveis da droga
Populações de Doentes Especiais • Doença Renal: metabolismo hepático igual, volume de distribuição igual/aumentado e eliminação prolongada intervalo doses • Doença Hepática: eliminação renal igual, volume de distribuição igual/aumentado, velocidade de metabolismo enzimático mais lento dosagem, intervalo doses
MEIA - VIDA • Não é um parâmetro farmacocinético primário, pois não pode der ligado apenas a um processo fisiológico. Ex: clearence (eliminação pelos órgãos) t½ = 0, 693. Vd Kel Cl Kel – constante de eliminação Vd – volume de distribuição Cl – clearence total
Equações utilizadas na farmacocinética dose única • Concentração plasmática a qualquer tempo após a administração C = S. F. D Vd (ng/ml, µg/ml, mg/ml)
Equações utilizadas na farmacocinética dose única • Eliminação: Clearence total (depuração plasmática) Cl. T = S. F. D Cl. T = Vd. K AUCT (ml/min, ml/min/Kg)
Equações utilizadas na farmacocinética dose única • Distribuição: volume aparente de distribuição Vd = S. F. D C (L, L/Kg) Vd = Cl. T K
Equações utilizadas na farmacocinética dose única • Eliminação Renal Fel = Ae Fração absoluta (µg, mg) Dose Ae (quantidade excretada) = Conv. x volume urina Cl. R = Fel. Cl. T Cl. NR = Cl. T – Cl. R (ml/min)
Princípios Farmacocinéticos • “Steady State”: a quantidade de droga administrada é igual à quantidade de droga eliminada dentro de um intervalo de doses, resultando num plateau ou nivel sérico da droga constante • Drogas com semi-vida curta atingem o “steady state” rapidamente; drogas com semi-vida longa demoram dias a semanas a atingir o “steady state”
SILVA, P. Farmacologia. 6 ed. Rio de janeiro: Guanabara Koogan, 2002. KATZUNG, B. G. Farmacologia Básica e Clínica. 9 ed. Rio de Janeiro: Guanabara Koogan, 2003 HARDMAN, j. g. , LIMBIRD, L. E. , MOLINOFF, P. R. , RUDDON, R. W. , GILMAN, A. G. The Pharmacological Basis of Therapeutics. 9 ed. New York: Mc Graw Hill. 1996 Chung Man Chin, Elizabeth Igne Ferreira Quím. Nova vol. 22 n. 1 São Paulo Jan. /Feb. Química Nova, 1999