COLECCIN FARMACOLOGIA SIMPLIFICADA UNIDAD 6 PRINCIPIOS DE FARMACOLOGIA





























- Slides: 33
“COLECCIÓN” FARMACOLOGIA SIMPLIFICADA UNIDAD 6 PRINCIPIOS DE FARMACOLOGIA FARMACOCINETICA: DISTRIBUCION DE FARMACOS Jorge Luis Maya Benavides Q. F. www. farmacus. com. co
CONTENIDO Unidad 6 6. 1. Las formas farmacéuticas y sus excipientes 1. 2. Componentes de los medicamentos 1. 3. Formas farmacéuticas 1. 4. Excipientes 1. 5. Clasificación de las formas farmacéuticas 1. 5. 1 Clasificación según el grado de esterilidad 1. 5. 2 Clasificación según el estado físico 1. 5. 3 Clasificación según la vía de administración 1. 6. Sistemas de liberación de fármacos 1. 6. 1 Formas farmacéuticas de liberación prolongada 1. 6. 2 Formas farmacéuticas de liberación retardada 1. 6. 3 Formas farmacéuticas de liberación en sitio especifico
Unidad 6 DISTRIBUCION DE FARMACOS
DISTRIBUCIÓN DEL AGUA EN EL ORGANISMO Se distinguen dos grandes compartimentos: intracelular y extracelular. 1. INTRACELULAR: está limitado por las membranas plasmáticas. Este compartimiento contiene dos tercios del agua corporal total, unos 27 litros en un varón adulto de 70 kg de peso. 2. EXTRACELULAR: Todas las células se encuentran inmersas en este líquido, de él reciben los nutrientes necesarios para sus procesos anabólicos (de síntesis) y en él vierten sus desechos catabólicos. Este comprende los líquidos intravascular e intersticial. a. El líquido intravascular o plasma sanguíneo se encuentra confinado en el sistema canalicular del aparato circulatorio. b. El líquido intersticial toma contacto directo con los cuerpos celulares, bañados por él. c. El liquido transcelular, que comprende el líquido contenido en la luz de los tractos digestivo, urinario y respiratorio; los líquidos cefalorraquídeo, pleural, pericárdico y sinovial y el humor acuoso del globo ocular A través de la pared de los capilares se realiza un activo intercambio entre los líquidos intravascular e intersticial En el espacio extracelular se encuentra un 20 por ciento del agua total, aproximadamente 12 litros en un adulto normal de 70 kilogramos.
POSIBILIDADES DE DISTRIBUCION DE UN FARMACO Persona de 70 KG de peso Plasma (3, 5 lt) + Agua Inters. (10, 5 LT)+Agua Intracelular(28 LT)=60% V=0, 6 L/Kg Plasma + Agua Intersticial=20% LEC V=0, 2 L/Kg
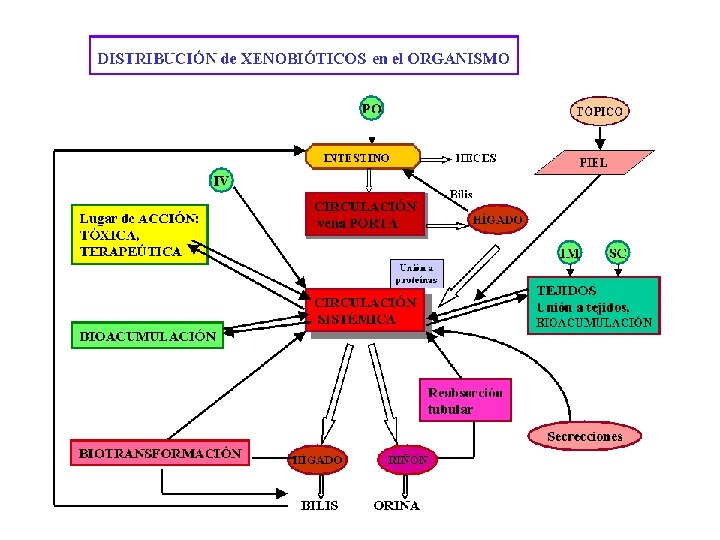
DISTRIBUCIÓN DE FARMACOS Después de que un fármaco se absorbe o se inyecta en el torrente circulatorio se distribuye en los líquidos intersticial y celular La Distribución es el reparto del fármaco por el organismo que permite su acceso a los diferentes órganos donde va actuar o va a ser eliminado. La distribución del fármaco condiciona las concentraciones que alcanzara en cada tejido en tal sentido que permite explicar el retraso en el comienzo de la acción o la finalización del efecto farmacológico de los fármacos. Los elementos que rigen la rapidez de llegada y la posible cantidad de fármaco que se distribuye en los tejidos son el gasto cardiaco, la corriente sanguínea regional y el volumen histico (tisular). 1. Primera fase: hígado, riñones, encéfalo son los que reciben la mayor cantidad de fármacos, en tanto que es mucho mas lenta la llegada a musculo, vísceras, piel y grasas. Esta fase de segunda distribución quizás necesite de minutos a horas para que la concentración de fármacos en los tejidos entre en una fase de equilibrio por distribución, con lo que hay en sangre. 2. Segunda fase: incluye una fracción mucho mayor de la masa corporal, que la fase inicial y por lo común explica gran parte de la distribución extravascular del fármaco. A excepción del encéfalo, la difusión del fármaco en el liquido intersticial es mas rápida por la naturaleza permeable del endotelio capilar.
DISTRIBUCION DE FARMACOS Tejidos altamente per fundidos Corazón Pulmones Sistema hepatoportal Cerebro Sistema espinal Tejidos escasamente per fundidos Tejidos de perfusión despreciable Musculo Piel Tejido adiposo Medula ósea Huesos Dientes Cartílagos Pelos LA DISTRIBUCION EN TEJIDOS VA DEPENDER DE FACTORES COMO a. Gradiente de p. H entre los líquidos intracelular y extracelular, en el caso de medicamentos que son ácidos o bases débiles b. Flujo sanguíneo regional c. Grado de unión del fármaco a proteínas plasmáticas d. Hidrofobia relativa del fármaco e. Volumen tisular
FACTORES QUE AFECTAN LA DISTRIBUCIÓN 1. LA UNIÓN A PROTEÍNAS PLASMÁTICAS 2. FIJACION A TEJIDOS 3. LA TASA DE EXTRACCIÓN 4. LOS VOLÚMENES FÍSICOS DEL ORGANISMO El fármaco en el plasma puede viajar de forma libre o unido a proteínas plasmáticas El factor determinante de mayor cuantía en la partición sangre/tejido es la unión del fármaco a proteínas plasmáticas. Solo la fracción de fármaco que no se encuentra unida a proteínas plasmáticas puede atravesar las membranas biológicas y en consecuencia puede: a. Unirse a los receptores celulares b. Distribuirse por los tejidos corporales c. Sufrir reacciones metabólicas d. Ser filtrada y excretada por el riñón
1. UNIÓN A PROTEÍNAS La eficacia de un fármaco puede ser afectada por el grado de unión a las proteínas dentro del plasma sanguíneo. Un fármaco en la sangre existe en dos formas ligado o suelto. Dependiendo de la afinidad específica del fármaco con el plasma, una proporción del mismo puede unirse a las proteínas del plasma y el resto quedar libre, si la interacción molecular proteínica es reversible, entonces existirá un equilibrio químico entre los estados libre y ligado, ej: Proteína + fármaco ⇌ Complejo Fàrmaco-Proteína Notablemente, es la fracción libre la que exhibe los efectos farmacológicos.
Es también la proporción libre la que puede ser metabolizada o excretada. Por ejemplo la fracción límite del anticoagulante warfarina es un 99%. Esto significa que la cantidad de Warfarina en la sangre un 99% está unida a las proteínas del plasma. El remanente 1% es la cantidad activa y que puede ser excretada. La proporción ligada a la proteína puede actuar como un depósito o reserva del fármaco que es liberado lentamente como proporción libre. Mientras que la parte libre es metabolizada o excretada del cuerpo, la proporción ligada será liberada a fin de mantener el equilibrio. Esto es importante a la hora de considerar las interacciones farmacológicas: un fármaco con un índice de fijación a proteínas plasmáticas inferior al 90%, si es desplazado de su unión a las proteínas por otro fármaco no va a aumentar significativamente su presencia en los tejidos. Por el contrario, con índices de unión a proteínas plasmáticas superiores al 95%, pequeños desplazamientos pueden originar importantes modificaciones de la concentración tisular y, por tanto, mayor riesgo de toxicidad por exceso de su efecto en los tejidos.
La unión a proteína aumenta la permanencia del fármaco en organismo. PRINCIPALES PROTEINAS PLASMATICAS La mas importante de todas es la albumina. 1. albumina(fármacos ácidos y neutros). Se unen salicilatos, AINES, penicilinas, sulfonamidas, anticoagulantes orales. Tiene 4 puntos de unión: a. Tipo I: Warfarina (poco especifico, se unen fármacos y la bilirrubina) b. Tipo II: Diazepam (mas especifico, se unen ácidos carboxílicos) Distintos sitios de unión para distintos c. Tipo III: Tamoxifeno fármacos en la Albumina d. Tipo IV: Digoxina 2. alfa 1 -glucoproteina acida (fármacos bases débiles) 3. lipoproteínas (bases débiles + fármacos liposolubles) 4. globulinas (trasporte de sustancias endógenas, ej. Esteroides, hormonas, vitaminas Antes de desplazo Después de desplazo % unido 95 90 % libre 5 10 % unido 50 45 % libre 50 55 % incremento de fracción libre Fármaco A +100 Fármaco B +10
UNION A PROTEINAS PLAMATICAS DE UN FARMACO Aquellos fármacos que se unen en gran proporción a proteínas plasmáticas tendrán mayor efecto farmacológico y mayor numero de efectos adversos. Ej. Fenitoina, diazepam, anticoagulantes orales Aquellos fármacos con índice terapéutico estrecho y se unen en gran proporción a proteínas plasmáticas, su desplazamiento presentara gran toxicidad. Ej. Anticoagulantes orales=crisis hemorrágicas Sulfonilureas= crisis hipoglucemias Bilirrubina= kernicterus neonatal
Dado que la albúmina es ligeramente básica, los fármacos ácidos o neutrales se unirán primariamente a ella. Si la albúmina se satura , entonces los fármacos se unirán a las lipoproteínas. Los fármacos de perfil básico se unirán a la ácida alfa 1 glicoproteína ácida. Esto es relevante ya que diversas condiciones médicas pueden afectar los niveles de proteínas plasmáticas. FACTORES QUE VARIAN LA UNION DEL FARMACO A PROTEINAS a. Hipoalbuminemias: la disminución de la cantidad de proteínas por a. Hipoalbuminemias: desnutrición o catabolismo hará aumentar los niveles plasmáticos de la forma libre y consecuentemente sus efectos. b. Situaciones clínicas: que modificación de los niveles de proteínas plasmáticas (hipoalbuminemias secundarias a procesos renales como el síndrome nefrótico o hepatopatías) pueden tener transcendencia en el efecto y toxicidad de un fármaco que presente índices de unión a proteínas plasmáticas superiores al 90% (ó 0, 9) disminuyen la unión y aumenta la fracción libre. Así como los trastornos como cáncer, artritis, infarto de miocardio, enfermedad de crohn, permiten que se incremente los valores de glucoproteina acida alfa y se genere una mayor union de fármacos alcalinos(básicos). Para fármacos con índice terapéutico estrecho algunas veces es preocupante el cambio en la concentración del fármaco libre inmediatamente después de administrar un fármaco que genere desplazamiento, como sucede con el anticoagulante warfarina
c. Interacción entre fármacos: Es la competencia por una misma proteína c. plasmática por varios fármacos. Usar dos fármacos al mismo tiempo puede afectar las fracciones libres respectivas de manera recíproca. Por ejemplo, asumimos que el fármaco A y B ambos son unibles a las proteínas. Si el A se administra, se unirá a las proteínas plasmáticas de la sangre. Si se administra el fármaco B, puede desplazar al A de las proteínas, incrementando la fracción libre de A, ello incrementa los efectos de A ya que solamente una fracción libre puede exhibir actividad. Ejemplo: Nótese que para el fármaco A, el % de incremento de la fracción libre es de un 100%, de ahí que el efecto farmacológico se ha duplicado. Este cambio en el efecto podría tener consecuencias adversas. Este efecto de la unión proteínica es muy significante con fármacos que son altamente unibles a las proteínas (>95%) y tienen un bajo índice terapéutico altamente unibles a las proteínas (>95%) y tienen un bajo (baja ventana terapéutica), tales como la warfarina. Un bajo índice terapéutico indica que hay un alto riesgo de toxicidad al usar el fármaco. En vista que la warfarina es un anticoagulante con un bajo índice terapéutico, la warfarina puede causar hemorragia si no se mantiene el grado de efecto farmacológico. Si un paciente que está bajo administración de warfarina e ingiere otro fármaco que pueda desplazar a la warfarina de la unión proteínica ello puede incrementar el riesgo de una hemorragia.
d. Alteración cuantitativa de la proteína: La modificación del sitio de unión con el fármaco dificulta su unión y aumenta los niveles de fármaco libre en sangre. e. Edad: Los Recién nacidos, tienen menor unión a proteínas plasmáticas, por lo que tendrán mayor fracción libre del fármaco. Los ancianos, tienen menor concentración de albumina y mayor de alfa 1 glucoproteina acida. f. La Gestación: Tienen menor unión a proteínas plasmáticas(albumina), por lo que tendrán mayor fracción libre del fármaco.
2. FIJACION A TEJIDOS Muchos fármacos se acumulan en los tejidos en concentraciones mayores que el liquido extracelular y sangre, de esta manera se reduce la acción del fármaco en su lugar de acción Ej. el antipalúdico quinacrina, la concentración en hígado es 100 mas que en sangre. la gentamicina se acumula en riñón y en sistema vestibular. La grasa como deposito: muchos fármacos liposolubles se almacenan por solución en la grasa neutra, esto es impórtate en personas obesas. Ej. El tiopental puede hallarse en la grasa corporal 3 horas después de administrado. El hueso sirve como acumulador de fármacos quelantes de iones metálicos divalentes como las tetraciclinas por adsorción a su trama cristalina, esto es importante en el tratamiento dela osteoporosis para el caso de los fosfonatos como el etidronato de sodio.
3. TASA DE EXTRACCIÓN Se refiere a la proporción del fármaco que es retirado de la circulación por cada órgano, una vez que el flujo sanguíneo lo haya hecho pasar a través de dicho órgano. Este nuevo concepto integra otros anteriores, ya que la tasa de extracción va a depender de distintos factores: a. Características del fármaco, entre ellas su p. Ka. b. Redistribución tisular: Por lo regular la terminación del efecto del fármaco ocurre por biotransformacion y excreción, pero eso también puede ser consecuencia de la redistribución desde el sitio de acción hacia otros tejidos o lugares. En algunos fármacos se produce una distribución rápida e intensa en determinados tejidos, hasta llegar al equilibrio con la concentración plasmática. Sin embargo otros tejidos más lentos continúan retirando fármaco del plasma, con lo que la concentración en el primer tejido queda por encima de la plasmática y por tanto sale fármaco del tejido hacia el plasma. Este fenómeno se sigue sucediendo durante un tiempo hasta alcanzar el equilibrio definitivo. Se obtiene por tanto dos concentraciones del fármaco en el tejido más sensible: una inicial más elevada y otra posterior consecuencia de la redistribución tisular. c. Diferencial de concentración con los tejidos. d. Superficie de intercambio
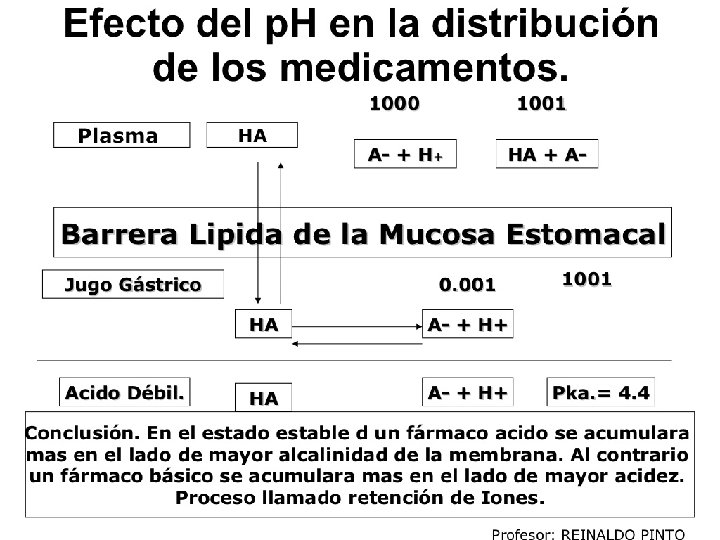
e. Presencia de barreras biológicas. Son obstáculos a la difusión similares a las encontradas en la absorción. Los fármacos de tamaño pequeño pasan por filtración (a feto, saliva, encéfalo y lágrimas no pasa). Ej. Barrera hematoencefálica: el fármaco puede llegar al S. N. C. por dos vías: Circulación capilar y liquido céfalo raquídeo, pero, la estructura del endotelio a nivel del SNC es diferente del endotelio vascular normal. En los endotelios normales, el fármaco puede pasar al espacio intersticial atravesando el endotelio vascular; en el cerebro no puede atravesar este espacio, porque el capilar está completamente cerrado y aísla el cerebro y el liquido cefalorraquídeo del resto del organismo; si la partícula es muy pequeña y liposoluble, como la heroína, el fármaco puede pasar de la sangre muy rápidamente, lo contrario ocurre con los fármacos ionizados e hidrosolubles. Cuando las meninges están inflamadas se facilita el paso, ej. penicilinas Ej. el antihistamínico loratadina alcanza una concentración encefálica mucho menor que la difenhidramina, por ello los menos efectos sedantes. Ej. Los organofosforados como los insecticidas por ser liposolubles pasan la barrera, y causan intoxicaciones. Las células de endotelio de capilares encefálicos muestran uniones estrechas y continuas y como consecuencia, la penetración de fármacos al tejido encefálico depende del trasporte transcelular y no del para celular
Barrera placentaria: en la mujer embarazada la placenta evita el paso de gran cantidad de fármacos, gérmenes, sustancias toxicas y antígenos al feto, que pudieran perjudicarle. La placenta se consideraba impermeable a fármacos, pero de hecho limita el paso de muchos fármacos pero no todos. Los liposolubles la atraviesan fácilmente pero no hay filtración mientras que los hidrosolubles y de peso molecular > 600 -1000 daltons no pasan. La permeabilidad crece sobre todo después del 3 er mes, con la disminución de espesor: 0, 02 mm al comienzo del embarazo hasta 0, 002 mm en las ultimas etapas de la gestación. El feto tiene menos proteínas plasmáticas, por tanto hay más fármaco libre y más efecto. El p. H es más ácido que el p. H maternal(7. 0 contra 7. 4), lo que modifica la absorción y distribución; los fármacos básicos se acumulan en el feto, y por tanto su concentración en el feto es superior que en la madre. Esa es la razón que nacen niños con síndrome de abstinencia de madres drogadictas.
Barrera hemato-testicular: Formada por las células de sertoli, que actúan a Barrera hemato-testicular: modo de barrera, impidiendo que cualquier sustancia pueda entrar en contacto con los espermatozoides y sus células precursoras. Barrera hemato-ocular: El ojo presenta las siguientes barreras que impiden la penetración de fármacos tanto instalados tópicamente(barrera corneal), como los aplicados sistémicamente(barrera hematoretiniana) Barrera hemato-prostatica: Son muy pocos los fármacos capaces de atravesar esta barrera en casos de infecciones prostáticas.
4. VOLÚMENES FÍSICOS DEL ORGANISMO Este concepto está relacionado con la multicompartimentalización. Considerando los fármacos como solutos, los distintos tejidos con especificidad del organismo van a actuar como los solventes que darán pie a las diferentes concentraciones del fármaco. Así, dependiendo de la naturaleza química de éste, habrá una especial predisposición de las sustancias liposolubles por la grasa corporal o de las hidrosolubles por el líquido extracelular. Este volumen de distribución (Vd) de un fármaco en el organismo es tan sólo aparente, no se refiere a un volumen fisiológico identificable, sino al volumen necesario para contener todo el fármaco en el cuerpo a las mismas concentraciones que esta presente en sangre o plasma. VDA= Cantidad de fármaco administrada_ Concentración alcanzada en plasma Un volumen de distribución bajo indica que el fármaco esta fuertemente unido a proteínas plasmáticas y no penetra en todos los compartimentos, indica que el fármaco esta confinado al compartimento sanguíneo o plasma, significa una pequeña distribución del fármaco en el organismo. Un volumen de distribución alto penetra en todos los compartimentos y la concentración del fármaco en los tejidos será alta y en el plasma será baja. Este concepto tiene interés clínico, pues a veces necesitamos alcanzar una determinada concentración de fármaco que sabemos es la óptima para que realice sus efectos en el organismo
Por ejemplo, si sabemos que la concentración terapéutica de un fármaco son 2, 5 mg/lt y que su volumen de distribución es de 100 lts. VDA= Cantidad de fármaco administrada_ Concentración alcanzada en plasma 100 lts= Cantidad de fármaco administrada 2, 5 mg/lt Cantidad de fármaco administrada= (100 lts)(2, 5 mg/lt)= 250 mgs La dosis inicial necesaria con la que se alcanzara la concentración eficaz =250 mgs Hay múltiples factores que pueden variar el volumen de distribución y por tanto es necesario hacer ajustes en la dosificación, tales como: a. Factores que alteren el volumen real, edemas, derrames, obesidad b. Factores que alteren la unión a proteínas
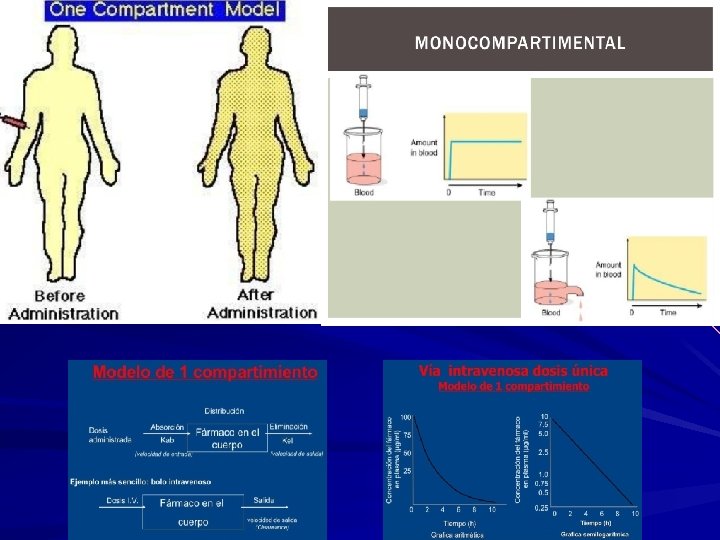
MODELOS DE DISTRIBUCION DE LOS FARMACOS Con el objetivo de simplificar los estudios de distribución se diseñaron modelos de funcionamiento basados fundamentalmente en la consideración del organismo compartimentos relacionados entre sí. Un compartimento es un volumen (teórico) en el cual el fármaco se encuentra distribuido uniformemente. Conceptualmente son mas fáciles de comprender. 1. Modelo monocompartimental considera el organismo, con la existencia de un solo compartimento. presupone que las concentraciones plasmáticas del fármaco son fiel reflejo de las concentraciones en otros fluidos o tejidos, y que la eliminación del fármaco es directamente proporcional a los niveles en el organismo del fármaco (cinética de primer grado). Este modelo asume que hay un solo compartimento: 1. El organismo=compartimento único 2. Distribucion de fármaco inmediata y homogénea 3. Liberacion y absorción se unen=entrada 4. Metabolismo y excreción se unen=eliminación 5. La entrada y eliminación del fármaco son directamente proporcionales a la concentración del fármaco
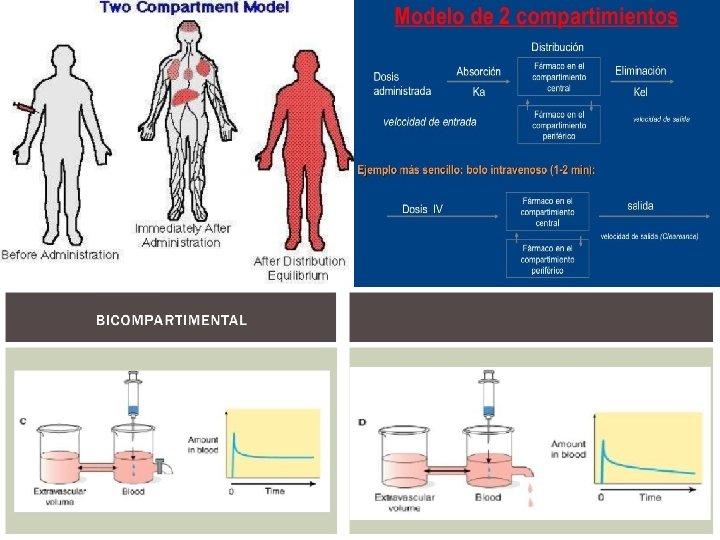
Sin embargo, no siempre este modelo recoge con una fidelidad aproximada lo que ocurre realmente en el organismo. Por ejemplo, no todos los tejidos presentan la misma riqueza en aporte sanguíneo, por lo que en unos la distribución del fármaco será más lenta que en otros. Además, existen algunos tejidos (como por ejemplo el tejido del cerebro) que presentan una verdadera barrera a la llegada de los fármacos, que será saltada con mayor o menor facilidad dependiendo de las características del fármaco. De modo que, manteniendo los otros condicionantes de proporcionalidad entre los distintos tejidos y de la velocidad de eliminación, el organismo se podría comportar como dos compartimentos: 2. Modelo bicompartimental, uno al que podemos llamar compartimento central que presenta una velocidad de distribución más elevada y constituido por los órganos y sistemas más intensamente irrigados y un compartimento periférico con una distribución mas lenta constituido por los órganos menos irrigados, quedando algunos tejidos como el cerebro en una posición variable según la facilidad que presente el fármaco para atravesar la barrera que lo separa de la sangre.
El modelo bicompartimental será diferente considerando en cual compartimento se produce la eliminación. Modelo un poco mas complejo basado en las siguientes premisas 1. Organismo= 2 compartimentos a. CENTRAL: órganos altamente irrigado o agua del organismo(riñón, hígado, pulmones) b. PERIFERICO: Órganos muy poco irrigados y tejidos(tejido adiposo, hueso) 2. Distribución en 2 fases a. CENTRAL: Rápida y homogénea, todo el agua b. PERIFERICA: Mas lenta, en tejidos 3. Liberación y absorción se unen=entrada 4. Metabolismo y excreción se unen=eliminación
No obstante, puede darse la situación de que la eliminación se realice desde el compartimento periférico o incluso desde ambos. Obtenemos así al menos tres variedades de modelo bicompartimental, que sin embargo sigue sin explicarnos todas las posibilidades. Además, la situación real es que cada tejido presenta sus propias características de distribución y que ninguna de ellas es estrictamente lineal. Se presenta así un modelo policomportamental, de numerosas curvas que precisaría complicadas ecuaciones para la obtención de una curva global. Por tanto, la elección del modelo va a depender de cuál sea el que ofrece el menor rango de error en función del tipo de fármaco implicado. La mayoría de los fármacos se distribuyen según el modelo bicompartimental. Los modelos de distribución ayudan a entender la velocidad de acción de los fármacos, así, cuando el efecto se produce en el compartimento central hay un paralelismo entre la concentración sanguínea y los efectos. Cuando el efecto se produce en el compartimento periférico hay una disociación entre las altas concentraciones plasmáticas y las bajas tisulares que ocasiona que los efectos sean mas retardados.
PARAMETROS FARMACOCINETICOS RELACIONADOS CON LA DISTRIBUCION 1. Volumen aparente de distribución 2. Porcentaje de unión a proteínas plasmáticas: oscila entre 0 hasta mas del 99%. Se puede considerar como una constante para cada fármaco, siendo independiente de la vía de administración y de su concentración plasmática.
BIBLIOGRAFIA Brunton, L; Lazo, J; Parker, K. Goodman & Gilman. Las bases farmacológicas de la terapéutica. Mc Graw Hill. 11 edición, 2007 http: //es. wikipedia. org/ Tratado de Fisiología Médica (Guyton y Hall) – Hall, John E. 12ª ed Color Atlas of Pharmacology. 2 nd edition, revised and expanded Heinz Lullmann, M. D. Professor Emeritus. Departament of Pharmacology University of Kiel Germany. Velasquez: Farmacología Básica y Clínica. P. Lorenzo, A. Moreno, J. C. Leza, I. Lizasoain y M. A. Moro. Editorial Panamericana. 18° Edición. www. facebook. com/farmacus. grupodecapacitacion Algunas imágenes publicadas han sido extraídas de internet por lo que damos nuestros mas sinceros agradecimientos a las fuentes. En todo caso no es nuestra intención atribuirse la autoría de ellas, solo se utilizan para crear un concepto grafico asociado al tema. www. farmacus. com. co Síguenos en twitter: @GRUPOFARMACUS Búscanos en Facebook: farmacus. grupodecapacitacion