Sequenze nucleotidiche Sequenze proteiche TRADUZIONE Esercizio relativo alla






 è una procedura per la predizione")


")




 Modellamento delle Regioni Loop Modellamento delle")














")





 può essere scaricato sul proprio computer")






- Slides: 51
Sequenze nucleotidiche Sequenze proteiche TRADUZIONE
Esercizio relativo alla traduzione di una sequenza nucleotidica in una sequenza amminoacidica Data la sequenza del gene della b-emoglobina umana: … ctggcccacaagtatcactac… 1)Scrivere la traduzione di questa sequenza in una sequenza amminoacidica 2)Scrivere la sequenza nucleotidica per un cambiamento di una singola base che produca una mutazione silente in questa regione (la mutazione silete è quella che lascia invariata la sequenza amminoacidica) 3)Scrivere la sequenza nucleotidica e la traduzione in sequenza amminoacidica per un cambiamento di una singola base che produca una mutazione di un amminoacido.
Ricerca di pattern e di motivi funzionali Qualche definizione necessaria ……. Un motivo di interesse biologico è costituito da un insieme di caratteri (nucleotidi o amminoacidi) non necessariamente contigui nella sequenza ma che si trovano sempre o sono spesso associati ad una precisa struttura e funzione biologica (ad esempio: promotori o hanno la stessa capacità di legare nucleotidi) La bioinformatica si occupa di sviluppare metodi per il riconoscimento di pattern di interesse biologico e di curare banche dati in cui tali pattern siano organizzati e resi disponibili per l’analisi strutturale e funzionale di nuove sequenze. Ciò deriva dal fatto che nel corso dell’evoluzione la natura ha sviluppato uno o pochi modi per erealizzare una nuova funzione (ad es. attività catalitica o altro)
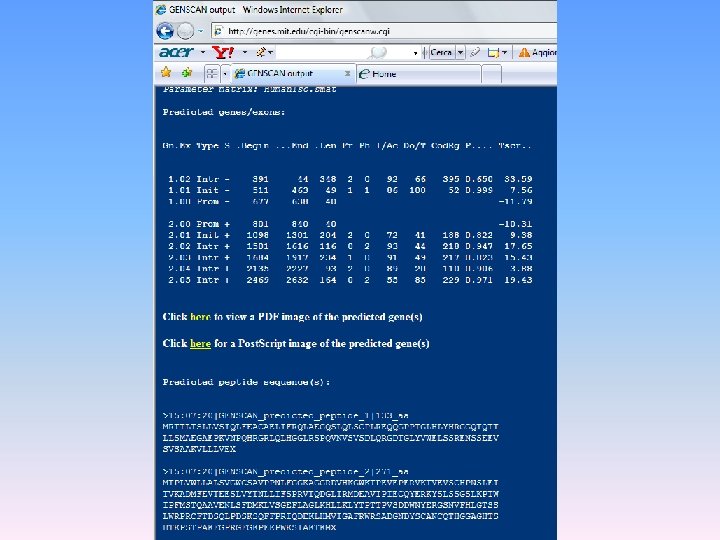
Per quanto riguarda la Ricerca di pattern e motivi funzionali in sequenze nucleotidiche Ricercare i siti di giunzione tra introni ed esoni Un gene è costituito da una sequenza codificante interrotta da sequenze non codificanti (dette introni). I geni sono combinazioni di corti esoni ed introni di lunghezza variabile. Il termine esoni si applica a tutte le regioni che non sono eliminate nel corso di maturazione del RNA [cioè le regioni non tradotte al 5’ dei geni, quelle codificanti vere e proprie (CDS) e le regioni non tradotte al 3’]. Identificare i siti di giunzione tra introni ed esoni per una corretta predizione della struttura di un gene. Net. Gene: http: //genome. cbs. dtu. dk/services/Net. Gene 2/ Gen. Scan: http: //genes. mit. edu/GENSCAN. html Gen. Scan è il programma più usato per predire la struttura di un gene
Esercizio 1: Predizioni dei geni codificanti proteine in sequenze genomiche mediante Gen. Scan Ricerchiamo i geni in una sequenza genomica prodotta nell’ambito del progetto di sequenziamento del genoma di Fugu. Collegandosi al sito: http: //fugu. hgmp. mrc. ac. uk/fugu-bin/clonesearch/ si effettua la ricerca della sequenza scaffold S 004519. La sequenza così estratta può essere utilizzata per la predizione utilizzando il programma Gen. Scan. La sequenza estratta viene incollata nella box clicca su Run Gen. Scan Nell’output di Gen. Scan sono indicati tutti i geni predetti, per ciascuno dei quali viene riportata la corrispondente ipotetica sequenza amminoacidica. Queste sequenze possono essere caratterizzati: o effettuando una ricerca con BLAST contro la banca dati delle proteine o ricercando i domini o motivi funzionali attraverso il sistema Inter. Pro
Esercizio 2: Stabilire con precisione la struttura di uno specifico gene usando Genome. Scan (http: //genes. mit. edu/genomescan. html) A partire dalla sequenza genomica (S 004519) e dalla proteina omologa selezionata con la più alta percentuale di identità di sequenza Run. Genome. Scan
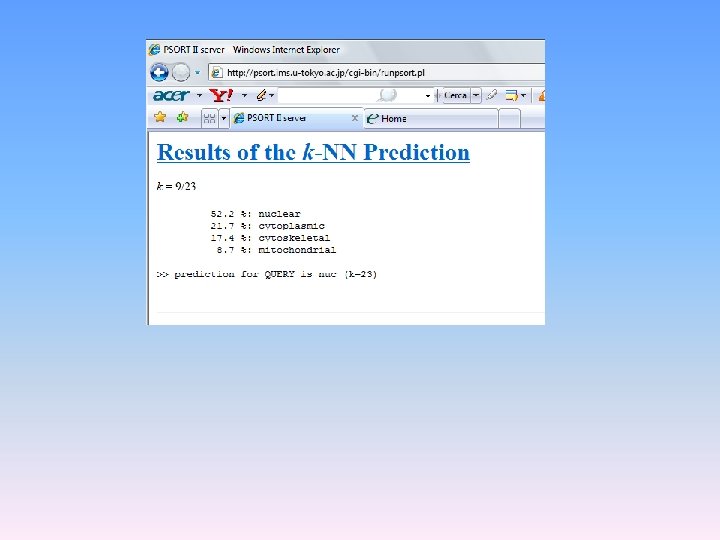
PSORT (http: //psort. nibb. ac. jp/form 2. html) è una procedura per la predizione della localizzazione delle proteine nella cellula. Riceve informazioni sottoforma di sequenze proteiche associate a localizzazioni subcellulari e ne ricava regole di associazione empiriche. Applicando queste regole ad una sequenza proteica di localizzazione ignota, PSORT giunge a predire la localizzazione, fornendo anche un indice di affidabilità della predizione.
Inserire Sequenza
Predizione della Struttura terziaria delle proteine
Funzione Strutture Sequenze From Tramontano (2004)
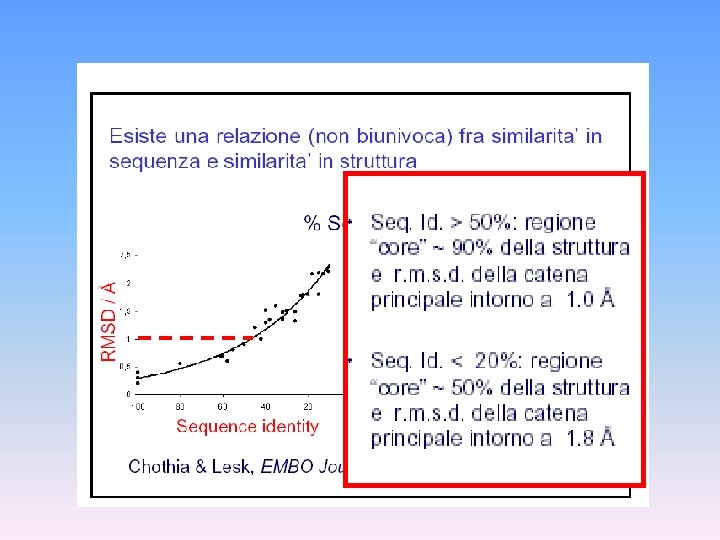
Predizione della struttura tridimensionale delle proteine Esistono proteine con sequenza simile e struttura 3 D nota ? NO Informazione minima necessaria: Sequenza della proteina SI Fold recognition ? La sequenza in esame è compatibile con una struttura 3 D nota? Modellamento per omologia Allineamento sequenze SI Costruzione del modello sul riferimento della struttura nota Verifica della qualità del modello NO Modellamento “ab initio” From Costantini et al. , 2006
Modellamento Comparativo SEQUENZA ………AQYSKRREVQCSVTDSEKRSLVLVPNSME LHAVMLQGGSDRCKVQL…… BLAST RICERCA DEL TEMPLATO CLUSTALW VALUTAZIONE DEL MODELLO TARGET-TEMPLATE PROSA MODELLO ALLINEAMENTO PROCHECK MODELLER
Modellamento Comparativo Modellamento delle Regioni strutturalmente conservate (SCR) Modellamento delle Regioni Loop Modellamento delle Catene Laterali Raffinamento del modellamento
Modellamento dei loop
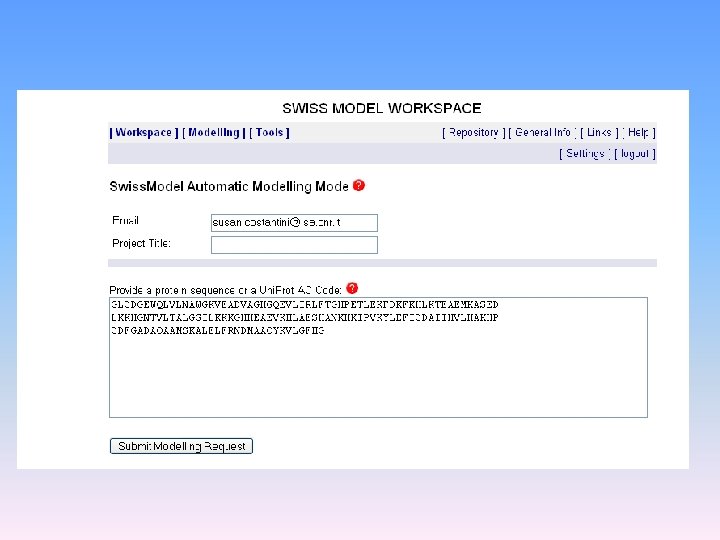
Modellamento per omologia SWISSMODELER Server automatici: SPARKS 2 SP 3 3 D-JIGSAW Metodi per valutare un modello ottenuto per omologia
Server che utilizza Modeller
3 D-JIGSAW
Metodi per valutare modelli di proteine http: //nihserver. mbi. ucla. edu/SAVS/
Per confrontare due strutture in termini di RMSD: http: //www. ebi. ac. uk/Dali. Lite/
PROSA: https: //prosa. services. came. sbg. ac. at/prosa. php
Fold Recognition
Fold noti……
Programmi per costruire modelli per riconoscimento del fold: 3 D-PSSM Based on sequence profiles, solvatation potentials and secondary structure http: //www. sbg. bio. ic. ac. uk/~3 dpssm/index 2. html FUGUE uses environment-based fold profiles that are created from structural alignments http: //www-cryst. bioc. cam. ac. uk/fugue/ SAMT 02 http: //www. soe. ucsc. edu/research/compbio/HMM-apps/T 02 -query. html The query is checked against a library of hidden Markov models. This is NOT a threading technique, it is sequence based. FFAS 03 (http: //ffas. ljcrf. edu/ffas-cgi/ffas. pl)This is a variant of FFAS, which aligns profile to profile. Profiles are generated differently from PSI-BLAST
THREADER può essere scaricato sul proprio computer
ANFINSEN Anfinsen ha dimostrato che alcune proteine in vitro possono essere sottoposte, introducendo agenti denaturanti quali la guanidina e l’urea, ad un processo reversibile di denaturazione, durante il quale perdono la loro struttura tridimensionale. Rimuovendo questi agenti denaturanti si riottiene la struttura tridimensionale attiva caratterizzata da una struttura tridimensionale compatta.
PARADOSSO DI LEVINTHAL Levinthal, che si pose il problema del tempo necessario affinché un sistema potesse raggiungere il suo stato di equilibrio. Infatti, supponendo che il numero di conformazioni accessibili al singolo amminoacido sia uguale a due (elica e foglietto beta), per una catena polipeptidica di 100 amminoacidi il numero totale di conformazioni possibili è 2100, che corrisponde a più di 1030. Se noi assumiamo che il tempo di interconversione da una conformazione alla sua alternativa è pari a 10 -11 secondi, il tempo necessario per una ricerca casuale di tutte le conformazioni è di 1011 anni. La soluzione di Levinthal a questo paradosso è stata che il meccanismo di folding è sottoposto ad un controllo di tipo cinetico, ovvero che esistono dei veri e propri percorsi definiti, che conducono dalla struttura casuale e lineare (U) alla struttura nativa e funzionale (N)
Robetta server (http: //robetta. bakerlab. org)
HMMSTR: http: //www. bioinfo. rpi. edu/~bystrc/hmmstr/server. php
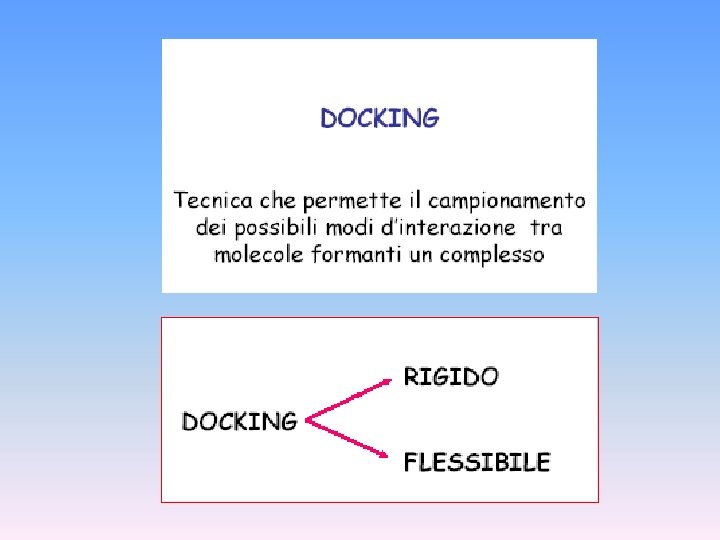
DOCKING RIGIDO DOCK AUTO-DOCK
DOCKING FLESSIBILE
Ci sono vari programmi di docking: Dock, Auto. Dock, Flex. X Questi programmi si basano sulla complementarietà geometrica e chimico-fisica tra le strutture, considerando spesso anche termini energetici ed entropici.
Clus. Pro: http: //nrc. bu. edu/cluster/
Z-Dock (http: //zlab. bu. edu/zdock/) può essere scaricato sul proprio computer
Z-Dock su server: http: //zdock. bu. edu/
http: //bioinfo 3 d. cs. tau. ac. il/Patch. Dock/
http: //vakser. bioinformatics. ku. edu/resources/grammx
http: //www. csd. abdn. ac. uk/hex_server/
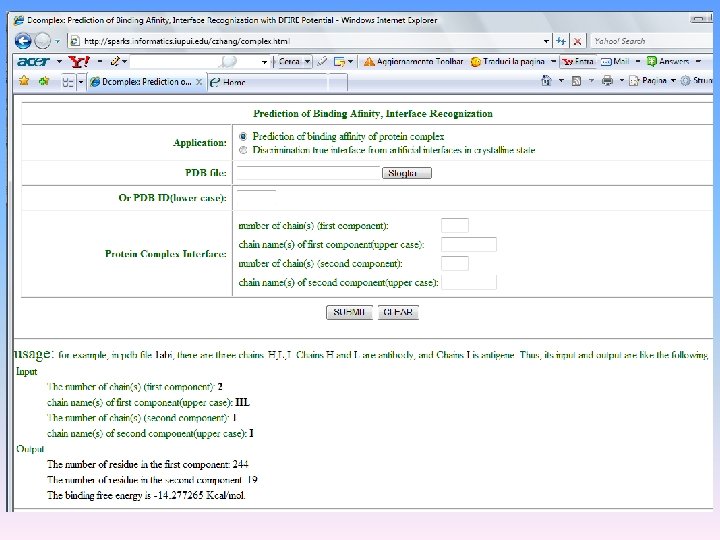
Protein Server valuta gli amminoacidi all’interfaccia Il programma DCOMPLEX valuta l’affinità di legame tra molecole complessate http: //sparks. informatics. iuoui. edu/czhang/complex. html
ESERCIZIO Prendendo come input il file del recettore e del ligando sul sito, provare ad eseguire un docking con i programmi visti prima.