IMMUNE DEFICIENCY DR REFIF ALSHAWK IMMUNODEFICIENCY DISEASE Caused








: • (phagocytes, neutrophils) • Defects are significant because of")

 • Defect in enzymes and microcidal molecules (NADPH oxidase; failure")
 • Defect in organelle membrane")
 • Absence of CD 18 – common βeta")

 Agammaglobulinemia • X-linked hyper-Ig. M syndrome")



 Agammaglobulinemia • Genetic disease that only")


 • Defect in gene encoding")







: • Due to defect in")



 • In about 50%")
 1. The x-linked SCID is due to a defect in")
 3. Mutations in the recombinase activating genes RAG-1 & RAG-2")
, the initial stages of hematopoietic cell development are blocked")











 • A defect in the autoimmune regulatory gene AIRE")
 SYNDROME • Patients with (IPEX) syndrome have inherited")
 SCID Common Variable Hypogglobulinemia/ X-linked")

- Slides: 56
IMMUNE DEFICIENCY DR. REFIF AL-SHAWK
• IMMUNODEFICIENCY DISEASE: Caused by defect in various components of immune system and results from a genetic or developmental defect or acquired factors in the immune system
IMMUNE DEFICIENCY OCCUR IN TWO DIFFERENT WAYS q PRIMARY I. D. -USUALLY GENETIC, CONGENITAL q SECONDARY I. D. -ACQUIRED (INFECTION , THERAPEUTIC TREATMENTS, CANCER & MALNUTRITION)
MECHANISMS OF IMMUNODEFICIENCY LOSS OR REDUCTION OF: • CELL TYPE • CELL NUMBERS • CELL FUNCTION
LOSS OF CELL FUNCTION • Receptors • Cell signaling • Cytokine production • Ig production • Co-stimulation impairment • Intracellular killing • Extravasation impairment
Characteristics seen in many immune deficiency diseases include the following: • Recurrent or chronic infections • Inability to clear infectious agents after standard antibiotic therapy • Unusual infectious agents
HEMATOPOIESIS
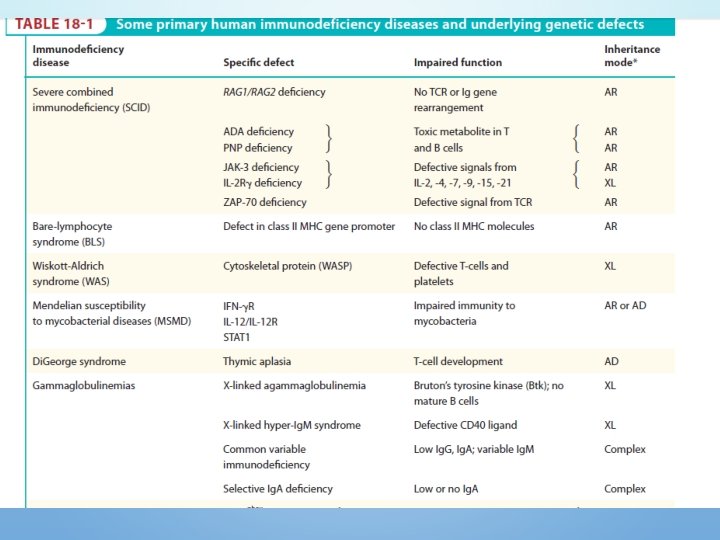
PRIMARY IMMUNODEFICIENCY • Myeloid lineage • Congenital agranulocytosis • Leukocyte-adhesion deficiency • Chediak-higashi syndrome • Lymphoid lineage • Severe combined immunodeficiency (SCID) • B cells • Agammaglobulinemia • Hypogammaglobulinemia • Specific Ig Deficiencies • T cells • Di. George Syndrome • Wiskott Aldrich Syndrome • Complement or its Regulation · Hereditary angoedema
DEFECT IN PHAGOCYTIC CELLS (MYELOID): • (phagocytes, neutrophils) • Defects are significant because of their key role in innate and adaptive I. R. • affect both ability to kill microb ( Chronic granulomatous disease, Chediak-higashi syndrome ) interactions with other cell (Leukocyte adhesion defect 1)
DEFECTS IN MYELOID LINEAGE Progenitor
Chronic Granulomatous Disease (CGD) • Defect in enzymes and microcidal molecules (NADPH oxidase; failure to generate superoxide anion & other O 2 radicals) • So the microorganisms will be ingested but not killed • Symptoms : recurrent infections with catalase- positive bacteria and fungi specially Staphylococcus aureus
Chediak- Higashi Syndrome • Normal levels of these enzymes(digestive) • Defect in organelle membrane which inhibits normal fusion of lysosomes • Fail to destroy ingested microbes • Symptoms : Recurrent infection with bacteria (chemotactic and degranulation defects, absent NK activity, partial albinism)
Leukocyte Adhesion Defect 1 ( LAD-1) • Absence of CD 18 – common βeta chain of leukocyte integrin, and become un able to migrate • Symptoms : Recurrent and chronic infection , fail to form pus
LYMPHOID LINEAGE • B cells • X-linked (Bruton) Agammaglobulinemia • X-linked hyper-Ig. M syndrome • Specific Ig Deficiencies (Selective Ig. A deficiency) • Common variable immunodeficiency • T cells • Di. George Syndrome • Wiskott Aldrich Syndrom • Bare Lymphocyte syndrome/ MHC II deficiency • MHC I deficiency • Complete functional B and T cell deficiency • Sever combined immunodeficiency (SCID)
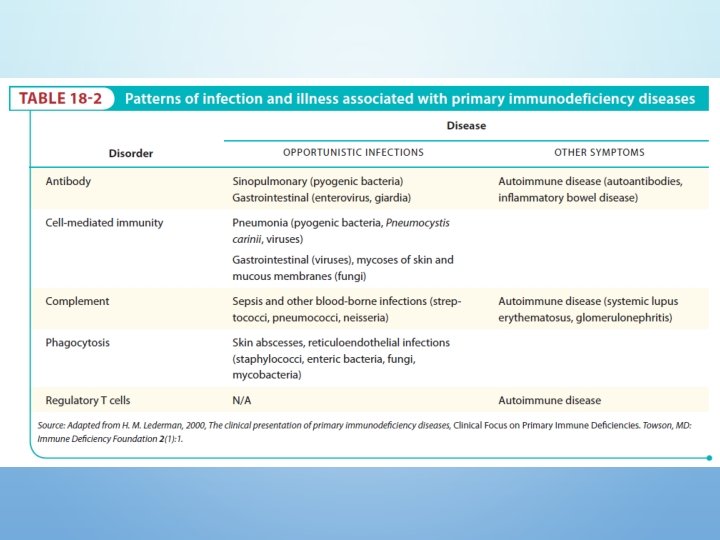
Defect In Humoral Immunity • These B-cell defects are responsible for the majority (more than 80%) of human immunodeficiency diseases • Patients with common defects in B-cell function have pyogenic infection : pneumania otitis media sinusitis
DEFECTS IN B-CELL FUNCTION DUE TO Early B cell maturation blocked Isotype switching dose not occur Terminal differentiati on of B cell blocked T-cell to B-cell is defective
DEFECTS IN B CELL DEVELOPMENT Progenitor
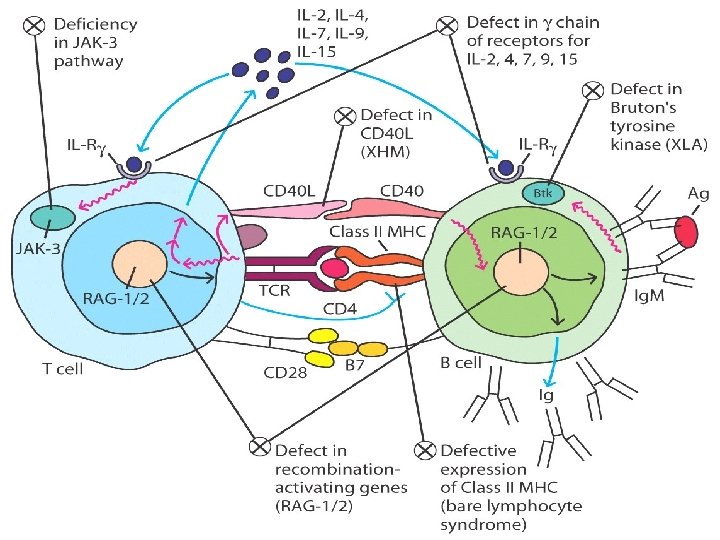
Early B Cell Maturation Fails In X-linked (Bruton) Agammaglobulinemia • Genetic disease that only affects males which results in few or no B cells in their blood or lymphoid tissue • The genetic defect in gene Bruton tyrosine kinase (BTK) an enzyme that is critical to early differentiation of B-cells into plasma cells which ultimately are responsible for producing immunoglobins • Because of abnormally low immunoglobin levels, the patient is susceptible to certain infections (encapsulated bact. ) such as: • Meningitis • Recurrent ear infections • Chronic sinus/upper respiratory infections • Treatment : monthly gammaglobulin replacement, antibiotics
Otitis media
Terminal Differentiation Of B Cells Fails In Selective Ig. A Deficiency • Most common immunoglobulin deficiency affecting 1 to 2 per thousand individuals • Main physical characteristic is a severe reduction in the levels of serum and secretory Ig. A that results from the inability of mature B cells to transform into Ig. A secreting plasma cells • Usually there are normal level of other isotypes • Persons with this disease tend to be affected with: • Chronic upper respiratory infections • Chronic GI infections • Increased incidence of allergic diseases such as asthma • Treatment : antibiotics, not immunoglobulins
Isotype Switching Dose Not Occur. . Hyper-Igm (HIg. M) • Defect in gene encoding the CD 40 Ligand on T cell which is important in the costimulatory signal required for class switching from Ig. M to Ig. G, Ig. A and Ig. E (The B-cell response to T-independent antigens only) • Symptoms : High serum titers of Ig. M without other isotypes , normal B & T cell • Although this form of immunodeficiency results in alterations in antibody production and presents with symptoms similar to HIM variants, it is classified as a CID • Increased susceptibility to extracellular bacteria & opportunists • Treatment : antibiotics and gammaglobulins
CD 40 LIGAND B Th CD 40 ligand Cytokines - IL-4, 5, 6
Examples for humoral immunity defects. Disease Bruton Xlinked hypogammag loulinemia Molecular defect Def of tyrosine kinase so blocks Bcell maturation Def of X-linked hyper-Ig. M CD 40 L on activated syndrome T cell Symptoms/signs 1. Low Ig of all classes. 2. No circulating B cell. 3. B-cell maturation stopped at pre-B stage. 4. Normal CMI. Treatment 1. Monthly gammaglobulin replacement. 2. Antibiotic. 1. Higher serum titer Antibiotic & of Ig. M only. 2. Normal gammaglobulin. B & T cell number. 3. Susceptibility to EC bacteria & opportunists.
Disease Molecular defect Symptoms/signs Treatment Selective Ig. A Deficiency Repeated Antibiotic, deficiency of Ig. A sinopulmonary & GIT not Ig. infections. Common Unknown 1. onset in late teens. Antibiotics variable 2. B cell present in immunodef peripheral blood. 3. Ig level decrease with time. 4. increase autoimmunity & atopy
T- CELL DEFICIENCY • Caused by problem with T lymphocytes (both CD 4+ helper cells and CD 8+ cytotoxic killer cells) • Patient’s have more severe symptoms than with humoral immunodeficiency
DEFECT IN T-CELL Combined partial B & T cell defect Defect in cells that are critical to the development or activation of T cell (APC) Reduced MHC I molecules Decrease No. of functional CD 8+ & NK Reduced MHC II molecules Decrease No. of functional CD 4+ Defects in thymic development (abnormal embryonic changes) Prevent thymic education of T cell
DEFECT IN T CELL DEVELOPMENT Progenitor
DEFECTS IN THYMIC DEVELOPMENT DIGEORGE'S SYNDROME: v. It is the most understood T-cell immunodeficienc v. Also known as congenital thymic aplasia/hypoplasia v. Associated with hypoparathyroidism, congenital heart disease, fish shaped mouth. v Defects results from abnormal development of fetus during 6 th-10 th week of gestation when parathyroid, thymus, lips, ears and aortic arch are being formed
MHC DEFICIENCY CLASS II DEFICIENCY (BARE LYMPHOCYTES SYNDROME II): • Due to defect in the MHC class II transactivator protein gene, which results in a lack of class-II MHC molecule on APC. • Patients have fewer CD 4 cells , immunoglobulin levels decreased owing to defective T-cell help • Increased susceptibility to infection
MHC DEFICIENCY CLASS I DEFICIENCY (BARE LYMPHOCYTES SYNDROME I OR TAP- 1 OR 2 DEFICIENCY): • There also individuals who have a defect in their transport associated protein (TAP) gene and hence do not express the class-I MHC molecules and consequently are deficient in CD 8+ T cells , CD 4+ normal • recurrent viral infection , normal DTH, normal Ab production
COMBIEND PARTIAL B- AND T-CELL DEFICIENCY ATAXIA-TELANGIECTASIA: • Defect in kinase involved in cell cycle • Associated with a lack of coordination of movement (ataxis) and dilation of small blood vessels of the facial area (telangiectasis). • T-cells and their functions are reduced to various degrees. • B cell numbers and Ig. M concentrations are normal to low.
DEFECTS IN LYMPHOID LINEAGE Progenitor
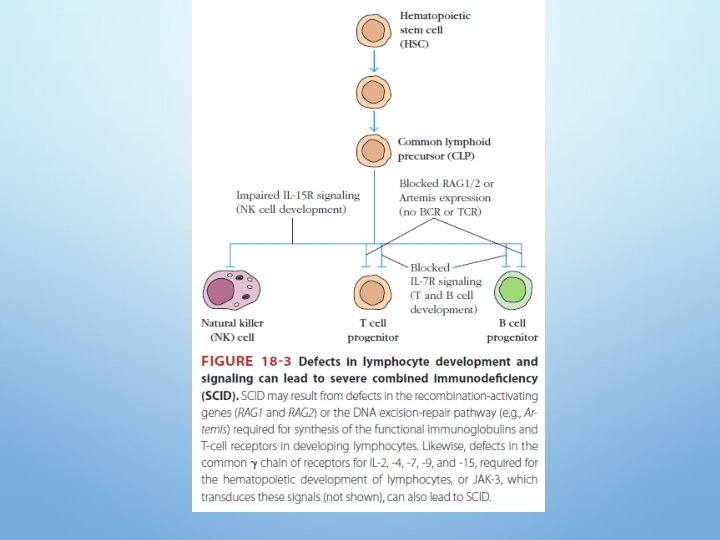
COMPLETE FUNCTIONAL B- AND TCELL DEFICIENCY SEVERE COMBINED IMMUNODEFICENCY (SCID) • In about 50% of SCID patients the immunodeficiency is x-linked whereas in the other half the deficiency is autosomal. • They are both characterized by an absence of T cell and B cell immunity and absence (or very low numbers) of circulating T and B lymphocytes. • Patients with SCID are susceptible to a variety of bacterial, viral, mycotic and protozoan infections.
SEVERE COMBINED IMMUNODEFICENCY (SCID) 1. The x-linked SCID is due to a defect in gammachain of IL-2 also shared by IL-4, -7, -11 and 15, all involved in lymphocyte proliferation and/or differentiation. (more common in male) 2. the autosomal SCIDs arise primarily from defects in adenosine deaminase (ADA) or purine nucleoside phosphorylase (PNP) genes which results is accumulation of d. ATP or d. GTP, respectively, and cause toxicity to lymphoid stem cells and apoptosis(T, B &NK)
SEVERE COMBINED IMMUNODEFICENCY (SCID) 3. Mutations in the recombinase activating genes RAG-1 & RAG-2 gene deficiency which are absolutely required for cleaving ds. DNA befor recombination of DNA to form the Ig genes encoding T cell receptors leading to undeveloped B & T cell 4. & genes encoding proteins involved in the DNA excisionrepair pathways employed during gene rearrangement (e. g. , Artemis) can also lead to SCID
5. In reticular dysgenesis (RD), the initial stages of hematopoietic cell development are blocked by defects in the adenylate kinase 2 gene (AK 2), favoring apoptosis of myeloid and lymphoid precursors and resulting in severe reductions in circulating leukocytes
screening test for SCID develop the standard blood samples collected from neonates via heel or finger pricks • Rapid polymerase chain reaction (PCR)-based assay looks for evidence of gene recombination as in excised DNA from the TCR or BCR locus, called T -cell receptor excision circles (TRECs) and -deleting recombination excision circles (KRECs). • In 2010, recommendations to screen every newborn for SCID were approved.
B and T-cell deficiency divided into these categories: A. Selective T-cell deficiency: Disease Defect Clinical manifestation Depression of T cell number with absence of responses. Di. George syndrome Thymic aplasia MHC class I deficiency Failure of TAP 1 1. CD 8+ T cell def. 2. CD 4+ T cell normal. molecule to 3. Recurrent viral infection. transport peptide to 4. Normal Ab formation. endoplasmic reticulum 1. def of CD 4+ T cell. Defects in transcription factors. 2. Hypogammagloulinemia. MHC class II def(Bare lymphocyte syndrome) 3. Clinically as SCID.
B. COMBINED PARTIAL B AND T-CELL DEFICIENCY: Ataxia telangiectasia Defect in kinase 1. gait abnormality. involved in the 2. Telangectasia (capillary distortion cell cycle. in the eye). 3. def of Ig. A & Ig. E production. C. Complete functional B and T cell deficiency: Sever combined Defects in common 1. Opportunistic (fungal) ID(SCID). γ chain of IL-2 infection. receptor. 2. Low level of circulating lymphocyte.
DISORDERS OF COMPLEMENT SYSTEM: • Complement abnormalities also lead to increased susceptibility to infections. • There are genetic deficiencies of various components of complement system, which lead to increased infections. • The most serious among these is the C 3 deficiency which may arise from low C 3 synthesis or deficiency in factor I or factor H.
DISORDERS OF COMPLEMENT SYSTEM DUE TO Classical pathway Both pathway Deficiencies in complement regulatory proteins
DEFECT IN CLASSICAL PATHWAY • Deficiencies of the classical pathway C 1 q, C 1 r, C 1 s, C 4, or C 2 • Result in a propensity to develop immune complex diseases such as SLE because it required for the dissolution of immune complexes • Increasing the risk of immune complex diseases SLE and increased infections with pyogenic bacteria
DEFICIENCY IN BOTH PATHWAYS • Deficiency in C 3 result in recurrent bacterial infection (pyogenic) indicating the importing role of C 3 in opsonization of pyogenic bacteria • Deficiency of the terminal components C 5 -C 8 result in remarkable susceptibility to infection with meningococcal & gonoccocal infections
DEFICIENCIES IN COMPLEMENT REGULATORY PROTEINS • The most important deficiency of the complement system is C 1 inhibitor which is responsible for dissociation of activated C 1 by binding to C 1 r 2 C 1 s 2 • Deficiency result in Hereditary angioedema (HAE) • Patients have recurrent Episodes of swelling at mucosal surfaces
4. DEFECTS OF COMPLEMENT. Deficiencies of complement or its regulation as in these cases: Components Deficiency Signs/diagnosis Classic pathway C 1 q, C 1 r, C 1 s, 1. Marked increase in immune C 4, C 2 complex disease. 2. Increased infection with pyogenic bacteria. Both pathways C 3 1. Recurrent bacterial infection. 2. Immune complex disease. C 5, C 6, C 7, C 8 Recurrent meningococcal & gonococcal infections. Def of regulatory C 1 -INH proteins. (hereditary angioedema) 1. Overuse of C 1, C 4 or C 2. 2. Edema at mucosal surfaces.
Type Of Infection Helps Predict The Type Of Immunodeficiency • B lymphocyte - pyogenic bacteria lungs • T lymphocyte - viruses, fungi, mycobacteria • Complement - meningococcus - CNS • Phagocyte - staphylococcus - skin
IMMUNODEFI CIENCY THAT DISRUPTS IMMUNE REGULATION CAN MANIFEST AS AUTOIMMUNITY • immune system must learn to recognize self. MHC proteins and to be proactive in suppressing reactions to self antigens in the host by the induction of tolerance in the thymus and by the surveillance activities of regulatory T cells. • Gene defect caused by inborn immunodeficiencies, actually manifest as immune overactivity, or autoimmunity
AUTOIMMUNE POLYENDOCRINOPATHY AND ECTODERMAL DYSTROPHY(APECED) • A defect in the autoimmune regulatory gene AIRE protein is expressed in medullary epithelial cells of the thymus, where it acts as a transcription factor to control expression of a whole host of tissuerestricted antigens.
IMMUNE DYSREGULATION, POLYENDOCRINOPATHY, ENTEROPATHY, X-LINKED (IPEX) SYNDROME • Patients with (IPEX) syndrome have inherited a mutated Fox. P 3 gene and lack expression of this protein, leading to a near absence of TREG cells. • Without these regulatory cells in the periphery, autoreactive T cells that have escaped central tolerance in the thymus go unchecked, leading to systemic autoimmune disease.
Overview of Primary Immunodeficiencies Di. George Syndrome (Autosomal dominant) SCID Common Variable Hypogglobulinemia/ X-linked hyper. Ig. M Syndrome/Selective Ig deficiency Progenitor x. LA Reticular Dysgenesis Progenitor Congenital Agranulocytosis Chronic Granulomatous Disease Auto. rec. /X-linked)
REFFERENCES : v IMMUNOLOGY , KUBY, SEVENTH EDITION 2013 v IMMUNOLOGY , KUBY, EIGHTH EDITION 2019 v CELLULAR AND MOLECULAR IMMUNOLOGY, ABUL K. ABBAS, 8 TH EDITION.