Estructura de gene procariota Promoter CDS UTR Terminator




")






")
 y")




")












; 5303 -5318")

 in human")
")


: • base composition • codon")

 for splice site, Kozak sequence or transcription factor")
20 -30 AAUAAA Stop codons UGA, UAG")










= 20 b=1 Alignment seq 1. pep seq 2.")



 Domain 2 (never found, inactive) Domain")



 PC=(0.")



 Gene. Machine http: //genome. nhgri. nih.")

 • Victor Solovyev and coleagues • FGENE applications are based on")

 • Combination of statistical methods (HMM) and neural networks • A candidate")



 • Genescan employs a dynamic programming strategy. •")

 • TWINSCAN models both gene structure and")


 • finding genes in microbial DNA. • combination of")
 to use bioinformatics tools • No single bioinformatics tool is 100 % accurate")






- Slides: 84
Estructura de gene procariota Promoter CDS UTR Terminator UTR Genomic DNA transcription m. RNA translation protein
Operons:
Prokaryotic Gene Organisation DNA Promoter Leader Repressor or Activator 5’ +1 Spacer m. RNA 3’ RNA Polymerase s Transcription: 2 consensus sequences and the startpoint - 10: TAATA T 80 A 95 t 45 A 60 a 50 T 96 - 35: TTGACA T 82 T 84 G 78 A 65 C 54 a 45 Translation: rbs (ribosomal binding site) Shine Delgarno AGGAGG Tailer Ribosome
promotores and reguladores en Procariotas • Promoter determines: 1. Which strand will serve as a template. 2. Transcription starting point. 3. Strength of polymerase binding. • RNA polymerase subunit for promoter recognition is called sigma-factor – – • • Different variations (7 for E. coli) Consensus binding sequences (Table 6. 2 in textbook) Operons for co-transcription Regulators affect the binding of RNA polymerase to DNA (positive and negative)
Ejemplo de promotor procariota • Pribnow box located at – 10 (6 -7 bp) • Promoter sequence located at -35 (6 bp)
Secuencias Consenso • Promoters sequences can vary tremendously. • RNA polymerase recognizes hundreds of different promoters
Terminadores • The terminator region pauses the polymerase and causes disassociation.
Producción de un ARN maduro en eucariotas • The final m. RNA may represent less than 5% of the transcribed DNA sequence
Modelo simplificado de un gen humano PROMOTOR Secuencia que no se traduce Intrón 1 Intrón 2 Secuencia que no se traduce Intrón 3 5` Región reguladora 3` EXON 1 EXON 2 EXON 3 EXON 4 EXON n Región reguladora Unidad de transcripción Después del procesamiento postranscripcional del ARN transcrito primario, la secuencia de ARNm corresponde a las secuencias de los exones y las no codificantes (intrones y UTRs).
Los genes eucariotas contienen normalmente intrones
Tipos de genes en eucariotas Protein encoding genes • Transcription • RNA Polymerase II dependent promoters • Type II splicing • Polyadenylation (exception histone m. RNAs) • Translation RNA coding genes • Transcription • RNA Polymerase I and III dependent promoters • Type I and III splicing • No polyadenylation • No translation
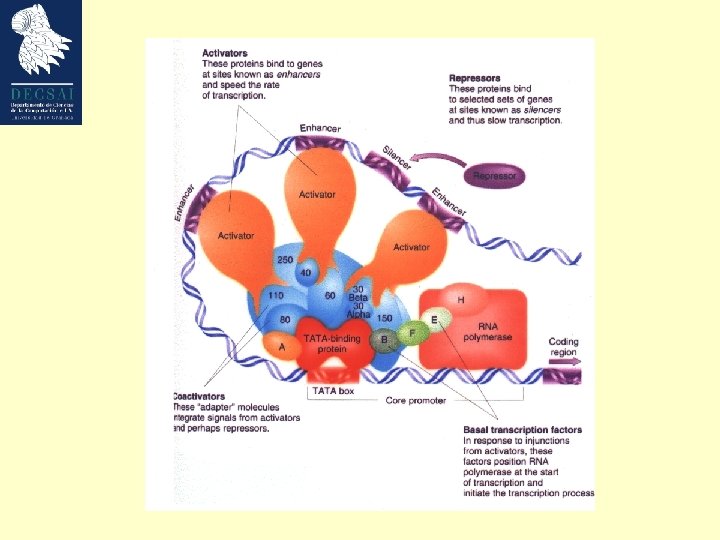
Estructura de un gen eucariota • TATA box located at – 25 – TATA(A/T) – Recognized by TATA-binding protein • Initiator sequence at +1 – YYCARR; Y is C/T, R is G/A – +1 is usually the A • Transcription factors bind to promoters – Position specific scoring matrix (PSSM) • Possible distant regions acting as enhancers or silencers (even more than 50 kb). – More complex mechanism than prokaryotes
La transcriptción puede ser modificada por factores que actuan en trans: activadores (enhancers) y silenciadores
El splicing alternativo puede producir diferentes proteinas con diferentes funciones Contains domains that adhere to cell surfaces Lacks domains that adhere to cell surfaces
Eukaryotic Gene Promoter GC CAAT Proximal Promoter proximal Organisation TSS TATAPromoter Inr Core core Transcription: core promoter: loosely conserved initiator region (Inr) around TSS ~ - 25: TATA-box proximal promoter: ~ - 75: CAT (CCAAT) ~ - 170: GC-box enhancer/silencer: upstream or downstream to promoter Translation: • 5‘ Kozak sequence: GCCACCATG • 3‘ polyadenylation site: AATAAA
Eukaryote gene structure vs. prokaryote gene structure • No operons • Capping at 5’ end and polyadenylation at 3’ end – Transport of m. RNA out of nucleus – Effects stability and efficiency of translation • Introns • Alternative splicing
Resumen • Prokaryotic genes promoter gene start gene terminator stop • Eukaryotic genes intron promoter exon start exon donor acceptor exon stop
Gene prediction: Prokaryotes vs. Eukaryotes Prokaryotes • Conserved promoter region (-10, -35; fixed spacing) • Contiguous open reading frames (ORF) • Polycistronic m. RNAs • Short intergenic sequences Good method: detecting large ORFs • Complications: • Sequencing errors • very small genes will be missed • Overlapping genes on both strands
Promoter and Gene prediction: Prokaryotes vs. Eukaryotes • Promoter elements • core promoter • initiator region (Inr) • TATA box • Downstream promoter element (DPE) • proximal promoter: transcription factor (“TF”) binding sites • CAAT box, • GC box • SP-1 sites • GAGA boxes • Enhancers/silencers sites (less useful) • Coding sequence • signal sensors (start and stop signals (Kozak sequence, stop codons), Polyadenylation signals, Splicing signals (3‘, 5‘ splice sites, splice junction, branchpoint) • content sensors (base composition, codon usage, hexamer usage)
El reto • The speed with which new data are collected increases and exceedes the rate with which they could be analysed. • Whole-genome sequences for more than 800 organisms (bacteria, archaea, and eukaryota as well as many viruses and organells) are either complete or being determined. • Across all sequenced species, nearly half of the potential genes can not be assigned a specific role.
Los programas para la predición de genes deberían ser capáces de identificar automáticamente y anotar todos los genes
Three Basic Strategies for Promoter and Gene Prediction • Búsqueda por homología • Análisis de señales en las secuencias • Análisis estadísticos
¿Porqué homología? Evolutionary relationships Paralogues: homologous proteins that perform different but related functions within one organism. ancestor Orthologues: homologous proteins that perform the same function in different species 1 species 2 species 3
Homology Searching • Investigate sequence databases such as EMBL or Swissprot with programs such as BLAST or TFASTA. • Orthologs / homologs / paralogs may have been described. Sequence identity may be low; several approaches should be tried. • As more sequence data is collected, this initial step becomes more important. Low coverage, high accuracy
Three Basic Strategies for Gene Prediction • Homology searching • Analysis of sequence signals • Statistical analysis
¿Que señales se pueden emplear en bioinformática para la predicción de genes?
¿Que diferencia a los genes de otras secuencias genómicas ? Genomic sequences tend towards randomness; Genes are non-random.
Base composition Translated DNA sequences are restricted in the choice for nucleotides in the first, second (and to a lesser extend) third position of the codons. Occurrence of a certain base in first, second and third position of the potential codons will not be random. 123123123123123123123123123 ATGATAGCTATACGGATCCGTAGCTAGATCAGTAGCGTGACTGCTGTCGTCATT A(1, 4, 7. . . )=10 of 18 (Random sequence Exp=25%) A(2, 5, 8. . . )=1 of 18 (Random sequence Exp=25%) Confidence levels can be calculated because large sets of coding and non-coding sequences have been analyzed.
Base composition bias Frequency of the four different nucleotides at the different codon positions in human coding regions.
Our model gene 1011 tata 1066 1345 2427 3058 Growth Factor Mouse Weakly expressed, tissue specific GC-rich (57%; cds 66%) ’TATA’ promoter (1011 -1017) 2 exons Not an easily predictable gene ! Bottner M, Laaff M, Schechinger B, Rappold G, Unsicker K, Suter-Crazzolara C. Gene. 1999 (237): 105 -11.
Testcode coding non-coding ‘Period three constraint’ [J. Fickett, Nucl. Acids Res. 10(17); 5303 -5318 (1982)]. The top and bottom regions predict coding and non-coding regions to a 95% confidence level. Start and stop codons (dashes and diamonds) are indicated.
Base composition bias Advantages: • Input: the crude DNA sequence • No information on reading frames is necessary. • No information on organism specific codon usage is needed. Disadvantages: • Short exons (<200 bp) are ignored. • Frameshift errors reduce the prediction success.
Codon usage bias The frequency of usage of each codon (per thousand) in human coding regions. The relative frequency of each codon among synonymous codons.
The human codon usage table (http: //www. kazusa. or. jp/codon/)
Codon usage bias Frequency of usage Leucine : Alanine : Tryptophan Protein encoding DNA = 6. 9 : 6. 5 : 1 Random DNA = 6. 0 : 4. 0 : 1 (Species specific, example rat) Relative Frequency Most amino acids are encoded by more than one codon. Leucine TTG TTA CTG CTA CTT human 12. 5 7. 2 40. 2 6. 9 12. 7 rat 12. 4 5. 0 40. 8 7. 0 11. 2 xenopus 14. 4 9. 1 26. 1 8. 4 15. 9 yeast 27. 1 26. 4 10. 4 13. 4 12. 2 Frequency dependent on species, level of gene expression. CTC 19. 4 20. 4 12. 6 5. 4
Codon usage bias Advantages: • Input: the crude DNA sequence AND a codon frequency table • No information on reading frame needed Disadvantages: • Weakly expressed genes have little bias • Frameshift errors reduce the prediction success
Analysis of Sequence Signals Content Sensors (Large sequence motifs): • base composition • codon usage • hexamer usage Signal Sensors (Short sequence motifs): • Start/stop codons • Splicing signals (3‘, 5‘ signals, branchpoint, splice junctions) • Polyadenylation signals • Transcription regulation signals (TF binding sites, promoters)
String matching Input: A text string t of length n. A patterns string p of length m. Output: All instances of the pattern in the text.
Patterns • Use consensus sequence (pattern) for splice site, Kozak sequence or transcription factor binding site. • Disadvantage: many false positives. TATA. . . ATGATATACAGATTATATAGATCGAT. . . TATA-box
Startcodon GCCACCAUGG Kozak sequence Polyadenylation signals YGUGUUYY (N)20 -30 AAUAAA Stop codons UGA, UAG Termination sequences (not well defined in eukaryotes)
Splice Sites 5 3 5 B B 5 5’ splice site CAG/GTAAGTAG 3 B 3 3’ splice site (T)10 NCAG/G(C) 9 B branchpoint CT(G/A)A(C/T) B J J + 3 3 J splicejunction MAG/G
Profile or Position Weigth Matrix • Replace the pattern by a profile • Employ training sets to build profile and to optimize the algorithm. Alignment 1234567. . . ACATTAA. . . TCAGAAT. . . ACAGAAC. . . AGATTAC. . . ACCGAAC. . . Profile A C G T consensus 1234567. . . 4040351. . . 0410003. . . 0103000. . . 1002201. . . ACAGAAC. . .
Three Basic Strategies for Gene Prediction • Homology searching • Analysis of sequence signals • Statistical analysis
Gribskov Profiles What is a Gribskov Profile? A Gribskov profile is a weight matrix of the probabilities of appearance of amino acids in a certain position in a multiple alignment. Score for finding each aa at a certain position POS 1 2 3 4 5 6 A -2 -2 18 -42 C 115 895 -65 -223 -104 -64 D -82 -302 -142 -62 -121 -221 E F -121 -401 -241 -81 -101 -161 -103 -283 -223 -163 -23 G 56 -304 416 196 -163 -223 H. . . L -101. . . -103 -302. . . -103 -221. . . -343 38. . . -302 -181. . . -43 -181. . . 176 . . . S T. . . Y . . . -101. . . -21 -61. . . -202. . . -101 102 -. . . -21 -282 181 -. . . 139 -162 81 218. . . -159 -182 -42. . . -121 -62 Gap 30 100 100 30
Gribskov Profiles Differences between Gribskov Profiles and common sequence comparison methods A group of related sequences can be used to build the profile The profile includes position-specific penalties for insertion and deletion
Gribskov Profiles What is needed to create a Gribskov Profile? A group of functionally related proteins Globins Immunoglobulins Aligned by seq 1. pep seq 2. pep seq 3. pep seq 4. pep seq 5. pep 1 ~CCGTL GCGSL~ ~CGHSV ~CGGTL CCGSS~ Similarity Three dimensional structure A mutational distance matrix Blosum 62 PAM 250 Dayhoff
Gribskov Profiles seq 1. pep seq 2. pep seq 3. pep seq 4. pep seq 5. pep Sequence position-specific scoring matrix M(p, a) 21 Columns 20 of them specify 1 specifies Score of each aa at a certain position Aligned positions A 1 2 3 4. . . N C D E . . . . W Y Penalty for deletion or insertion in that position Number of positions in the alignment Gap 1 ~CCGTL GCGSL~ ~CGHSV ~CGGTL CCGSS~
Creating a Gribskov Profile The profile is filled using the Multiple alignment Mutational distance matrix 20 M(p, a)= b=1 W(p, b) * Y(a, b) W(p, b) = n(b, p)/ NR Weight of appearance of aa b at position p n(b, p) is the number of times that aa b appears in position p NR number of rows in the alignment Y(a, b) Value in the mutational distance matrix M(p, C)= W(p, W) * Y(C, W)
Mutational Distance Matrix Blosum 62 matrix A B C D E F G H I K L M N P Q R S T V W X Y Z W A 4 -2 0 -2 -1 -1 -2 -1 -1 -1 1 0 0 -3 -1 -2 -1 B CC D E F G H I K L M N P Q R S T V W X Y Z 6 -3 6 2 -3 -1 -1 -3 -1 -4 -3 1 -1 0 -2 0 -1 -3 -4 -1 -3 2 9 -3 -4 -2 -3 -3 -1 -1 -1 -2 -4 6 2 -3 -1 -1 -3 -1 -4 -3 1 -1 0 -2 0 -1 -3 -4 -1 -3 2 5 -3 -2 0 -3 1 -3 -2 0 -1 2 0 0 -1 -2 -3 -1 -2 5 6 -3 -1 0 -3 0 0 -3 -4 -3 -3 -2 -2 -1 1 -1 3 -3 6 -2 -4 -3 0 -2 -2 -2 0 -2 -3 -2 -1 -3 -2 8 -3 -1 -3 -2 1 -2 0 0 -1 -2 -3 -2 -1 2 0 4 -3 2 1 -3 -3 -2 -1 3 -3 -1 -1 -3 5 -2 -1 0 -1 1 2 0 -1 -2 -3 -1 -2 1 4 2 -3 -3 -2 -2 -2 -1 1 -2 -1 -1 -3 5 -2 -2 0 -1 -1 -2 6 -2 0 0 1 0 -3 -4 -1 -2 0 7 -1 -2 -1 -1 -2 -4 -1 -3 -1 5 1 0 -1 -2 -2 -1 -1 2 5 -1 -1 -3 -3 -1 -2 0 4 1 -2 -3 -1 -2 0 5 0 -2 -1 4 -3 -1 -1 -2 11 -1 2 -3 -1 -1 -1 7 -2 5 -2
Creating a Gribskov Profile The profile is filled using the Multiple alignment Mutational distance matrix 20 M(p, a)= b=1 W(p, b) * Y(a, b) W(p, b) = n(b, p)/ NR Weight of appearance of aa b at position p n(b, p) is the number of times that aa b appears in position p NR number of rows in the alignment Y(a, b) Value in the mutational distance matrix M(p, C)= W(p, W) * Y(C, W) = -2
Creating a Gribskov Profile M(p, a)= 20 b=1 Alignment seq 1. pep seq 2. pep seq 3. pep seq 4. pep seq 5. pep W(p, b) * Y(a, b) M(1, A)= b=1 W(1, b) * Y(A, b) M(1, A)= ( W(1, A) * Y(A, A) ) + (W(1, C) * Y(A, C) ) +. . . + ( W(1, Y) *Y(A, Y) ) 1 ~CCGTL GCGSL~ ~CGHSV ~CGGTL CCGSS~ M(1, A)= ( 0. 025/6 * 4) + ( 1/6 * 0 ) +. . . + ( 0. 025/6 * -1) aa not present in a position get a very small weight 0, 025/NR M(1, C)= b=1 W(1, b) * Y(C, b) Consensus sequence symbol with largest value in each position (CCGGTL) Score for finding each aa at a certain position POS 1 2 3 4 5 6 A -2 -2 -2 18 -42 C 115 895 -65 -223 -104 -64 D -82 -302 -142 -62 -121 -221 E F -121 -401 -241 -81 -101 -161 -103 -283 -223 -163 -23 G 56 -304 416 196 -163 -223 H. . . L -101. . . -103 -302. . . -103 -221. . . -343 38. . . -302 -181. . . -43 -181. . . 176 . . . S T. . . Y . . . -101. . . -21 -61. . . -202. . . -101 102 -. . . -21 -282 181 -. . . 139 -162 81 218. . . -159 -182 -42. . . -121 -62 Gap 30 100 100 30
Scoring with a Gribskov Profile Alignment seq 1. pep seq 2. pep seq 3. pep seq 4. pep seq 5. pep Consensus sequence symbol with largest value in each position (CCGGTL) 1 ~CCGTL GCGSL~ ~CGHSV ~CGGTL CCGSS~ P(CCGGTL)= Pp 1(C)* Pp 2(C)* Pp 3(G)* Pp 4(G)* Pp 5(T) * Pp 6(L) P(CCGGTL)= log Pp 1(C)+ log Pp 2(C)+ log Pp 3(G)+ log Pp 4(G)+ log Pp 5(T)+ log Pp 6(L) Probability of any sequence is calculated in the same way Score for finding each aa at a certain position POS 1 2 3 4 5 6 A -2 -2 -2 18 -42 C 115 895 -65 -223 -104 -64 D -82 -302 -142 -62 -121 -221 E F -121 -401 -241 -81 -101 -161 -103 -283 -223 -163 -23 G 56 -304 416 196 -163 -223 H. . . L -101. . . -103 -302. . . -103 -221. . . -343 38. . . -302 -181. . . -43 -181. . . 176 . . . S T. . . Y . . . -101. . . -21 -61. . . -202. . . -101 102 -. . . -21 -282 181 -. . . 139 -162 81 218. . . -159 -182 -42. . . -121 -62 Gap 30 100 100 30
Introduction Gribskov Profile Definition Creating a Gribskov Profile Scoring a sequence with a Profile Hidden Markov Models Definition State order of an HMM Basic Architecture Scoring a sequence with an HMM Building a Hidden Markov Model Estimation of the model Problems building an HMM Biological application of HMMs HMM programs in HUSAR
Advantages of using Markov Models A P=0. 6 Markov Models are probabilistic, models, with a solid statistical foundation In contrast to patterns and profiles, HMMs allow consistent treatment of insertions and deletions P=0. 1 P=0. 2 C P=0. 09 P=0. 01 T G C - In contrast to patterns and profiles, Markov Models take into account the information about neighboring residues.
Hidden Markov Model Domain 1 (active binding site) Domain 2 (never found, inactive) Domain 3 (never found, inactive) Domain 4 (active) 123456… ATGTCGTCGTCG ATGTGGTCGTCG ATGTCATCGTCG ATGTGATCGTCG Markov Model is based on active domains only !! If a G is found at position 3, P(T)4=1. 0 If a T is found at position 4, P(C)5=0. 5, P(G)5=0. 5 If a C is found at position 5, P(A)6=0. 0, P(G)6=1. 0 If a G is found at position 5, P(A)6=1. 0, P(G)6=0. 0 If a G is found at position 6, P(T)7=1. 0 If an A is found at position 6, P(T)7=1. 0
Order state of HMMs Markov Models take into account additional information about neighboring residues. First order Markov Model Captures the first order correlation between neighboring nucleotides HMM models can use preceding, succeeding or surrounding residues Fifth order Markov Model There is no real limit in the number of preceding residues that can be used for an HMM (computing time!)
Biological applications of HMMs Gene finding Protein secondary structure prediction Protein homology recognition Phylogenetic analysis Radiation hybrid mapping Profile HMM libraries Genetic linkage mapping (Birney & Durbin, 1997; Henderson, 1997; Krogh, 1997; Lukashin & Borodovsky, 1998) (Goldman et al. , 1996) (Karplus et al. , 1999) (Felsenstein & Churchill, 1996) (Sloniw et al. , 1997) (PROSITE; Pfam database) (Krushyak et al. , 1996)
Hidden Markov Model transitions states t 1, 1 t t 1, 2 A 2, 2 t B P 1(a) P 2(a) P 1(b) P 2(b) a b a 2, end End HMM Observed symbol sequence P(aba|HMM) =P 1(a) t 1, 1 P 1(b) t 1, 2 P 2(a) t 2, end
Hidden Markov Model transitions states 0. 9 1. 0 Start E PA=(0. 25) PC=(0. 25) PG=(0. 25) PT=(0. 25) 0. 1 1. 0 5 I PA=(0. 05) PC=(0) PG=(0. 95) PT=(0) PA=(0. 4) PC=(0. 1) PG=(0. 1) PT=(0. 4) 0. 1 End
Hidden Markov Models assume that sequences are generated independently of the model Applied to time series or to linear sequences
Basic Architecture of a profile HMM 0. 3 d 1 Start d 2 d 3 End 0. 06 i 1 i 0 Probabilities m 1 C from information contained in Alignment A C 0. 01 D E F G H I. . . Y 0. 015 i 3 i 2 m 2 C A C D E F G H I. . . Y 0. 5 m 3 Y A C D E F G H I. . . Y Match states Model the distribution of symbols in the corresponding column of an alignment 0. 01
Methods in Gene Prediction Ab initio analysis of genomic sequences: Genscan (Burge and Karlin 1997) HMMer (Haussler et al. 1993, Krogh et al. 1994) FGenes. H (Solovyev and Salamov 1994) Comparison of protein and genomic sequences: Procrustes (Gelfand et al. 1996) Genewise (Birney and Durbin) Cross-species genomic sequence comparisons: CEM (Bafna and Huson 2000) TWINSCAN (Korf et al. 2001) Doublescan Meyer and Durbin 2002) SLAM (Alexandersson et al. 2003)
Gene prediction programs (many with homology searching capabilities) Gene. Machine http: //genome. nhgri. nih. gov/genemachine Genscan http: //genome. dkfz-heidelberg. de Genome. Scan http: //genes. mit. edu/genomescan Fgenesh, Fgenes-M, TSSW, TSSG, Polyah, SPL and RNAS http: //genomic. sanger. ac. uk/gf/gf. shtml Fgenesh, Fgenes-M, SPL and RNASPL http: //www. softberry. com/berry. phtml HMMgene http: //www. cbs. dtu. dk/services/HMMgene Genie http: //www. fruitfly. org/seq_tools/genie. html Gene. Mark http: //www. ebi. ac. uk/genemark Gene. ID http: //www 1. imim. es/software/geneid. html#top Gene. Parser http: //beagle. colorado. edu/~eesnyder/Gene. Parser. html MZEF and POMBE http: //argon. cshl. org/genefinder/ AAT, MZEF with homology http: //genome. cs. mtu. edu/aat. html MZEF with Splice. Proximal. Check http: //industry. ebi. ac. uk/~thanaraj/MZEF-SPC. html Genesplicer, Glimmer and Glimmer. M http: //www. tigr. org/~salzberg Web. Gene http: //www. itba. mi. cnr. it/webgene Gen. Lang http: //www. cbil. upenn. edu/genlang_home. html Xpound ftp: //igs-server. cnrs-mrs. fr/pub/Banbury/xpound Gene-prediction programs: alignment based Procrustes http: //www-hto. usc. edu/software/procrustes/index. hl Gene. Wise 2 http: //www. sanger. ac. uk/Software/Wise 2 Splice. Predictor http: //bioinformatics. iastate. edu/cgi-bin/sp. cgi Predict. Genes http: //cbrg. inf. ethz. ch/Server/subsection 3_1_8. html
Gene-prediction programs: comparative genomics Doublescan http: //www. sanger. ac. uk/Software/analysis/doublescan SLAM http: //bio. math. berkeley. edu/slam Twinscan http: / genes. cs. wustl. edu Finding ORFs and splice sites Dio. Genes http: //www. cbc. umn. edu/diogenes/index. html Orf. Finder http: //www. ncbi. nlm. nih. gov/gorf. html Yeast. Gene http: //tubic. tju. edu. cn/cgi-bin/Yeastgene. cgi CDS: search coding regions http: //bioweb. pasteur. fr/seqanal/interfaces/cds-simple. html Neural network splice site prediction http: //www. fruitfly. org/seq_tools/splice. html Net. Gene 2 http: //www. cbs. dtu. dk/services/Net. Gene 2 RNA gene prediction t. RNAScan http: //www. genetics. wustl. edu/eddy/t. RNAscan-SE/
FGENES, FGENEH, FGENESH(+) • Victor Solovyev and coleagues • FGENE applications are based on HMMs • They form a complete, partially automated, modular package • Dynamic modelling with various features of coding sequences • Precise determination of exon borders with homology search
1011 tata 1066 1345 2427 3058
GENIE (UCLA) • Combination of statistical methods (HMM) and neural networks • A candidate sequence is "threaded" through the HMM using a min-cost path search algorithm and the system reports this "optimal" path as the predicted gene structure.
1011 tata 1066 1345 2427 3058
Grail. EXP • Widely used for genbank annotations • Grail. EXP predicts exons, genes, promoters, poly. As, Cp. G islands, EST similarities, and repetitive elements
1011 tata 1066 1345 2427 3058
Genscan (Chris Burge and Samuel Karlin) • Genescan employs a dynamic programming strategy. • General three-periodic (inhomogeneous) fifth order Markov Model. • Transcription-, translation- and splicing signals. • Length distributions and compositional features of introns, exons and intergenic regions. • Exceptional: It was developed to recognize partial and multiple genes on both strands. • Independent of databases.
1011 tata 1066 1345 2427 3058
TWINSCAN (I. Korf et al. , 2001) • TWINSCAN models both gene structure and evolutionary conservation • Scores of features (e. g. splice sites, coding regions) are modified using the patterns of divergence between the target genome and a closely related genome.
Prediction of a subsequence of the mouse genome alignments to human genomic sequences repeat sequences reported TWINSCAN GENSCAN actual gene structure
1011 tata 1066 1345 2427 3058 c. DNA protein
GLIMMER (Salzberg and colleagues, JHU) • finding genes in microbial DNA. • combination of Markov models from first through eighth order, weighting each model according to its predictive power. • Widely used for genbank annotations.
How(not) to use bioinformatics tools • No single bioinformatics tool is 100 % accurate (colleagues and developers may tell you the opposite). • Common pitfall: for which organism was the application developed? • Repetitive elements (such as the mouse L 1 element) can be wrongly recognized as genes. • Bioinformatics rule: try several approaches, try to understand why they may give apparently contradicting results.
Evaluation of Gene Prediction Tools The ideal testset is a segment of DNA for which all genes have been described experimentally. Gene prediction tool Specificity = true predicted / all predicted Measure for false positives: 9 / 11 = 81. 8% Sensitivity = true predicted / true genes Measure for false negatives: 9 / 10 = 90%
Accuracy versus G+C content 1, 00 0, 90 0, 80 0, 70 Accuracy 0, 60 0 - 40% 40 - 50% 50 - 60% 60 - 100% 0, 50 0, 40 0, 30 0, 20 0, 10 0, 00 FGENES Gene. Mark Genie Genscan Morgan http: //www. cse. ucsc. edu/~rogic/evaluation/tablesgen. html MZEF
Exon accuracy 1, 00 0, 90 0, 80 Exon accuracy 0, 70 0, 60 Sensitivity (false negatives) 0, 50 Specificity (false positives) 0, 40 Partially correct predicted 0, 30 0, 20 0, 10 0, 00 FGENES Gene. Mark Genie Genscan HMMgene Morgan http: //www. cse. ucsc. edu/~rogic/evaluation/tablesgen. html MZEF
Accuracy versus exon length 1, 00 0, 90 0, 80 0, 70 0 - 24 25 - 49 50 - 74 75 - 99 100 - 199 200 - 299 300 + Accuracy 0, 60 0, 50 0, 40 0, 30 0, 20 0, 10 0, 00 FGENES Gene. Mark Genie Genscan HMMgene Morgan http: //www. cse. ucsc. edu/~rogic/evaluation/tablesgen. html MZEF
Accuracy versus exon type http: //www. cse. ucsc. edu/~rogic/evaluation/tablesgen. html
Open Problems and Future Directions • Near the 90% of the nucleotides can be identified correctly, but exact boundaries of the exons and their assemblies into complete coding sequences are much more difficult to predict. Less than the half of the genes are predicted exactly correct. • Multiple protein products correspond to a single gene through alternative splicing, alternative transcription or alternative translation has not been dealt with effectively. • Promoter recognition