Use of glucose as an energy source in










 AMP Gº′ = -41.")
 Adenine Phosphate groups Ribose 13")
 to pyruvate is catalyzed by pyruvate")



 - TCA plays several roles in metabolism, its")





 is present in")





 and (1→ 6) Glycosidic Bonds 1 4 (1→ 4) glycosidic bond 1")












 2 - Pomp’s disease")
 G 6 Pase catalyzes the final step")

 (α-1, 4 -glucosidase deficiency) This is the most")
 Amylo-1, 6 -glucosidase (debranching enzyme) deficiency In this")
 Amylo-(1, 4→ 1, 6)-Transglycosylase (branching enzyme) deficiency")
 Muscle phosphorylase deficiency Its major symptom, that")
 muscle phosphofrucktokinase deficiency The result of a deficiency")

 This is only disease of glycogen")
























 are an important source of energy for")




, intermediate density lipoproteins (IDL), and low density")








- Slides: 99
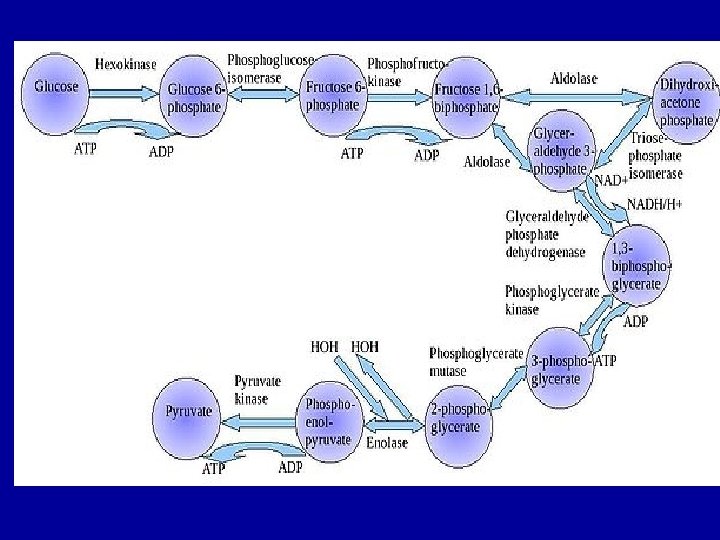
- Use of glucose as an energy source in cells is via aerobic or anaerobic respiration - Both of these start with the early steps of the glycolysis metabolic pathway - The first step of this is the phosphorylation of glucose by hexokinase to prepare it for later breakdown to provide energy. - The major reason for the immediate phosphorylation of glucose by a hexokinase is to prevent diffusion out of the cell. The phosphorylation adds a charged phosphate group so the glucose 6 -phosphate cannot easily cross the cell membrane. - Irreversible first steps of a metabolic pathway are common for regulatory purposes.
- In anaerobic respiration one glucose molecule produces a net gain of 2 ATP molecules (4 ATP molecules are produced during glycolysis but 2 are required by enzymes used during the process) - In aerobic respiration a molecule of glucose is much more profitable in that a net worth of 32 ATP molecules are generated (34 gross with 2 being required in the process)
- Second step, G 6 P is then rearranged into fructose 6 phosphate (F 6 P) by glucose phosphate isomerase - Fructose can also enter the glycolytic pathway by phosphorylation at this point. The reaction requires an enzyme, phosphohexose isomerase, to proceed. - This reaction is freely reversible under normal cell conditions. However, it is often driven forward because of a low concentration of F 6 P, which is constantly consumed during the next step of glycolysis. Under conditions of high F 6 P concentration, this reaction readily runs in reverse.
- Step three, Fructose 1, 6 -phophate is then split into two 3 -carbon sugars, namely dihydroxyaceton phosphate and glyceraldehyde 3 -phosphate. These 2 substances [triose phosphates] are biochemically interconvertible. - Step four, conversion of glyceraldehyde 3 -phosphate into 1, 3 -bisphoglycerate (1, 3 -BPG) by enzyme glyceraldyhade phophate dehydrogenase. - Step five, generation of ATP from 1, 3 -BPG by the enzyme phoshoglycerate kinase. - step six, generation of additional ATP and the formation of pyruvate, enzyme that catalyzed this step is pyruvate kinase.
- In glycolysis a molecule of glucose is degraded in a series of enzyme-catalyzed reactions to yeild 2 molecules of pyruvate. The three irreversible enzymecatalyzed reactions are: 1) Hexokinase (or glucokinase) 2) Phosphofructokinase 3) Pyruvate kinase Pyruvate then passes into the mitochondrion and enter krebs cycle
Metabolism: Major Metabolic Pathways 7
Hexokinase - Mammals have several isozymes of hexokinase that catalyze the phosphorylation of glucose to glucose 6 -phosphate - In most tissues the phosphorylation of glucose is catalyzed by hexokinase, that is one of the three regulatory enzymes of glycolysis. It has a broad specificity and is able to phosphorylate several hexoses in addition to glycosis. - Hexokinase is inhibited by the reaction product, glycose 6 -phosphate, that accumulates when further metabolism of this hexose phosphate is reduced, for example by a high ATP/ADP ratio. - Hexokinase has a high affinity for glucose. This permits efficient phosphorylation and subsequent metabolism of glucose even when tissue concentration of glucose are low.
Glucokinase occur in the liver and B-cells of the pancreas. It is the predominant enzyme for the phosphorylation of glucose. Glucokinase differs from hexokinase in requiring a much higher glucose concentration for half saturation. Therefore, glucokinase function only when the intracellular concentration of glucose in the hepatocyte is elevated such as during the brief period following consumption of a carbohydrate-rich meal, when high levels of glucose are delivered to the liver via portal vein. Glucokinase has a high Vmax allowing the liver to effectively remove this flood of glucose from the portal blood. This prevents large amount of glucose from entering the systemic circulation and thus minimizes hyperglycemia during the absorptive period.
Glucokinase levels are increased by carbohydrate-rich diets and by insulin. Glucokinase is not inhibited by glucose 6 -phosphate but hexokinase is inhibited by G 6 P. Regulation of Glycolysis (phosphofructokinase) - The main regulatory enzyme of the glycolytic pathway is phosphofructokinase that catalyzed the irreversible phosphrylation reaction. Other enzymes of the pathway can exhibit regulatory behavior. - Phosphofructokinase catalyzes a physiologically irreversible reaction, and this enzyme-catalyzed reaction represents the committed step in the pathway. - Phosphofructokinase activity is regulated by several allosteric effectors, such as ATP, AMP, citrate, and fructose 2, 6 -bisphosphate.
ATP and AMP ATP is both a substrate and an allosteric inhibitor of the phosphofructokinase reaction. When ATP concentrations are high, there is no need to accelerate glucose catabolism. Under condition in which the levels of ATP are decreased, there is an associate increase in ADP and AMP. Under these physiological conditions, AMP serves as an allosteric activator of phophofructokinase. Activation of phosphofructokinase promotes glycolysis and the generation of ATP, this occurs with the decrease in the concentration of AMP that, in turn, decreases the allosteric activation of phosphofructokinase. Regulation by AMP is important in exercising skeletal muscle but probably is not important in other cells.
How It Happens – ATP Hydrolysis + Inorganic pyrophosphate (PPi) AMP Gº′ = -41. 8 k. J mol-1 12
ATP (adenosine triphosphate) Adenine Phosphate groups Ribose 13
Formation of pyruvate The conversion of phosphoenolpyruvate (PEP) to pyruvate is catalyzed by pyruvate kinase, the third irreversible reaction of glycolysis In liver, pyruvate kinase is activated by fructose 1, 6 -bisphosphate, the product of the phosphofructokinase reaction. Phosphorylation by c. AMP-dependent protein kinase leads to inactivation of pyruvate kinase in the liver When blood glucose levels are low, elevated glucagons thus increases the intracellular level of c. AMP, that favors the phosphorylation and inactivation of pyruvate kinase
Carboxylation of pyruvate - The first step in the synthesis of glucose from pyruvate is the irreversible conversion of pyruvate to phosphoenolpyruvate (PEP) by pyruvate kinase. - In gluconeogenesis, pyruvate is first carboxylated by pyruvate carboxylase to oxaloacetate (OAA), that is then converted to PEP by the action of PEP-carboxykinase [Note that pyruvate carboxylase is also found in the mitochondria of liver and kidney cells, but not of muscle].
- Several vitamins provide cofactors for the enzymes involved in these metabolic pathways. E. g. the pyruvate dehydrogenase reaction need cofactors derived from thiamine, riboflavin, lipoic acid and pantothenic acid a deficiency of any of these could cause malfunctioning of a metabolic pathway at the particular enzymetic reaction where the cofactor is involved.
gluconeogenesis provides a substantial fraction of the glucose produced in fasting human even after a few hours fast. Gluconeogenesis occurs in liver and to a smaller extent in kidney. The noncarbohydrate precursors that can be converted to glucose include the glycolysis products lactate and pyruvate, citric acid cycle intermediates, and the carbon skeletons of most amino acids. However, all these substances must be converted to oxaloacetate, the starting material for gluconeogenesis.
Citric acid cycle TCA (Krebs cycle) - TCA plays several roles in metabolism, its central function is the oxidation of acetyl Co. A to CO 2 and H 2 O. - Acetyl Co. A is derived from the metabolism of fuel molecule such as amino acids, fatty acids, and carbohydrates. - The cycle occurs totally in the mitochondrial matrix and is therefore in close proximity to reactions of oxidative phosphorylation.
Regulation of the citric acid cycle - The pyruvate dehydrogenase complex that consist of multiple copies of each of the three enzymes: Pyruvate dehydrogenase (E 1) Dihydrlipoyl transacetylase (E 2) Dihydrolipoyl dehydrogenase (E 3) - Is regulated both allosterically and by covalent modification. The compex is strongly inhibited by ATP, as well as by acetyl-Co. A and NADH, the products of the reaction.
- The allosteric inhibition of pyruvate oxidation is greatly enhanced when long-chain fatty acids are available. - AMP, Co. A, and NAD+, all of which accumulate when too little acetate flows into TCA cycle, allosterically activate the pyruvate dehydrogenase complex. - Therefore this enzyme activity turned off when ample fuel is available in the form of fatty acids and acetyl. Co. A and when the cell’s ATP concentration and [NADH]/[NAD+] ratio are high. - And turned on when energy demands are high and greater flux of acetyl-Co. A into the TCA cycle is requierd.
In the pyruvate dehydrogenase complex these allosteric regulation mechanisms are complemented by a second level of regulation, covalent protein modification. Covalent modification: The pyruvate dehydrogenase complex exists in two forms: an active, nonphosphorylated form and an inactive, phophorylated form. Phophorylated and nonphosphorylated can be intercinverted by two separate enzymes, a kinase and a phosphatase. The kinase is activated by an increase in the ratio of acetyl. Co. A/Co. A or NADH/NAD+.
- An increase in the ratio of ADP/ATP, which signals increased demand for energy production, inhibits the kinase and allows the phosphatase to produce more of the active, nonphosphorylated enzyme. Regulation of the TCA cycle by activation and inhibition of enzyme activities - In contrast to glycolysis, which is regulated primarily by phosphofructokinase, the TCA cycle is controlled by regulation of several enzyme activities. - The most important of these regulated enzymes are citrate synthase, isocitrate dehydrogenase, and alpha-ketoglutarate dehydrogenase complex
Regulation by the availability of ADP: 1 - effects of elevated ADP: - Energy consumption due to muscular contraction, biosynthetic reactions, or other processes result in the hydrolysis of ATP to ADP and Pi. - The resulting increase in the concentration of ADP accelerates the rate of reaction that use ADP to generate ATP. - Production of ATP increases until it matches the rate of ATP consumption by energy-requiring reactions.
2 - effect of low ADP: - If ADP (or Pi) is present in limiting concentration, the formation of ATP by oxidative phosphorylation decreases due to lack of phosphate acceptor (ADP) or inorganic phosphate (Pi). - The rate of oxidative phosphorylation is proportinal to [ADP][Pi]/[ATP]. This is known as respiratory control of energy production. - The oxidation of NADH and FADH 2 by the respiratory chain also decline if ADP is limiting. This is because the processes of oxidation and phosphorylation are tightly coupled and must occur at once. - As NADH and FADH 2 accumulate, their oxidized forms become depleted, causing the oxidation of acteyl-Co. A by TCA cycle to be inhibited due to a lack of oxidized coenzymes.
The Reactions Involved 25
The Molecules Involved 26
Coenzyme A Reactive group 27
Glycosides Methyl -D-Glucopyranoside Glycosidic bond Methyl -D-Glucopyranoside 28
Disaccharides Are Glycosides 29
(1→ 4) and (1→ 6) Glycosidic Bonds 1 4 (1→ 4) glycosidic bond 1 6 (1→ 6) glycosidic bond 30
Polysaccharides 31
Glycogen Is A Polysaccharide 32
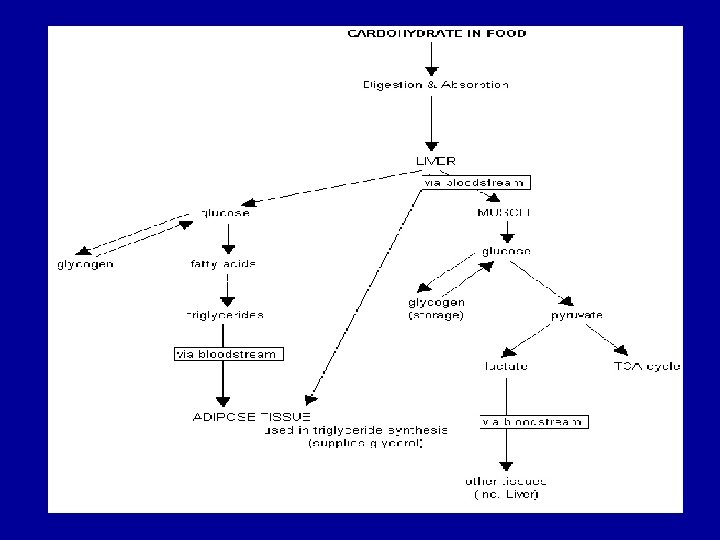
Glycogen metabolism Blood glucose can be obtained from 3 primary sources: - Diet - Degradation of glycogen - Gluconeogenesis In the absence of a dietary source of glucose, glucose is rapidly released from liver glycogen, similarly, muscle glycogen is extensively degraded in working muscle. When glycogen stores are depleted, specific tissues synthesize glucose de novo, using amino acids from the body proteins as the primary source of carbons for the gluconeogenic pathway.
Glycogen breakdown - In muscle, the need for ATP results in the conversion ogf glycogen to glucose 6 -phosphate (G 6 P) for entry into glycolysis. - In liver, low blood glucose concentration triggers glycogen breakdown to G 6 P, which in this case is hydrolyzed to glucose and released into the bloodstream to reverse this situation. - Glycogen breakdown requires the action of three enzymes: 1 - Glycogen phosphorylase catalyzes glycogen phosphorolysis (bond cleavage by the substitution of a phosphate group) to yield glucose 1 -phosphate (G 6 P). Glycogen + Pi (n residues) glycogen + G 1 P (n-1 residues) This enzyme will only release a glucose unit that is at least five units from a branch point.
2 - Glycogen debranching enzyme removes glycogen’s branches, thereby permitting the glycogen phosphotylase reaction to go to completion. It also hydrolyzes alpha(1 -6)-linked glucosyl units to yield glucose. Consequently, ~92% of glycogen’s glucose residues are converted to G 1 P. The remaining ~8%, those at the branch points, are converted to glucose. 3 - Phosphoglucomutae converts G 1 P to G 6 P, which is also formed in the first step of glycolysis through the action of either hexokinase or glucokinase. G 6 P can either continue along the glycolytic pathway (as in muscle) or be hydrolyzed to glucose (as in liver).
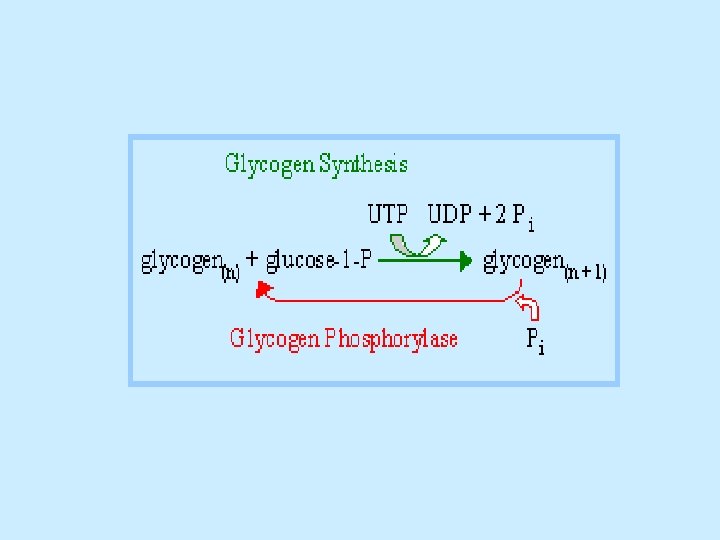
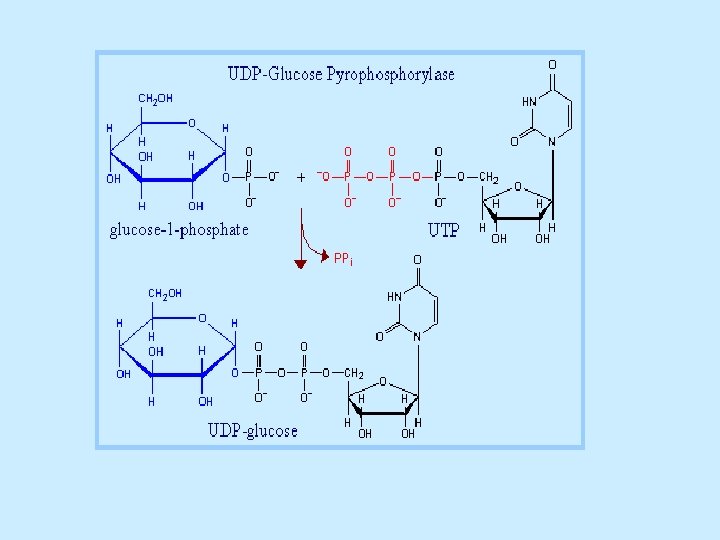
Glycogen synthesis Since the direct conversion og G 1 P to glycogen and Pi is thermodynamically unfavorable (positive ∆G) under all physiological Pi concentrations, glycogen biosynthesis requires an additional exergonic step. Luis Leloir 1957, combined G 1 P with uridine triphosphate (UTP) to form uridine diphosphate glucose (UDP-glucose or UDPG). The enzymes catalyzing the three steps involved in the glycogen synthesis pathway are: UDP-glucose pyrophosphorylase, glycogen synthase, glycogen branching enzyme. 1 - UDP-glucose pyrophosphorylase catalyzes the reaction of UTP and G 1 P. Phosphoryl oxygen og G 1 P attacks the alpha phosphorus atom of UTP to form UDPG and release PPi
2 - Glycogen synthase, the glucosyl unit of UDPG is transferred to the C 4 OH group on one of glycogen’s nonreducing ends to form an alpha(1 -4) glycosidic bond. The glycogen synthase reaction, like those of glycogen phophorylase and lysozyme, is though to involve a glucosyl oxonium ion intermediate or transition state. 3 - Glycogen branching, glycogen synthase catalyzes only α(1 -4) -linkage formation to yield α-amylose. Branching to form glycogen is accomplished by a separate enzyme, amylo-(1, 4 -1, 6)-transglycosylase (branching enzyme), which is distinct from glycogen debranching enzyme. Branches are created by transfer of terminal chain segments consisting of ~7 glucosyl residues to the C 6 -OH groups of glucose residues on the same or another glycogen chain. Each transferred segment must come from a chain of at least 11 residues, and the new branch point must be at least 4 residues away from other branch points.
Summary of glycogen metabolism - In the well-fed state, glycogen synthase is allosterically activated by glucose 6 -phosphate when it is present in elevated concentrations. In contrast, glycogen phophorylase is allosterically inhibited by glucose 6 phosphate, as well as by ATP, a high energy signal in the cell [ Note: in the liver, glucose also serves as an allosteric inhibitor of glycogen phosphorylase]. - Most of the glycogen molecule is degraded to glycose 1 -phosphate by the action of glycogen phosphorylase, the key enzyme in glycogen breakdown. The glycosidic linkage between C-1 of a terminal residue and C-4 of the adjacent one is split by orthophosphate to give glucose 1 -phosphate that can converted into glucose 6 phosphate.
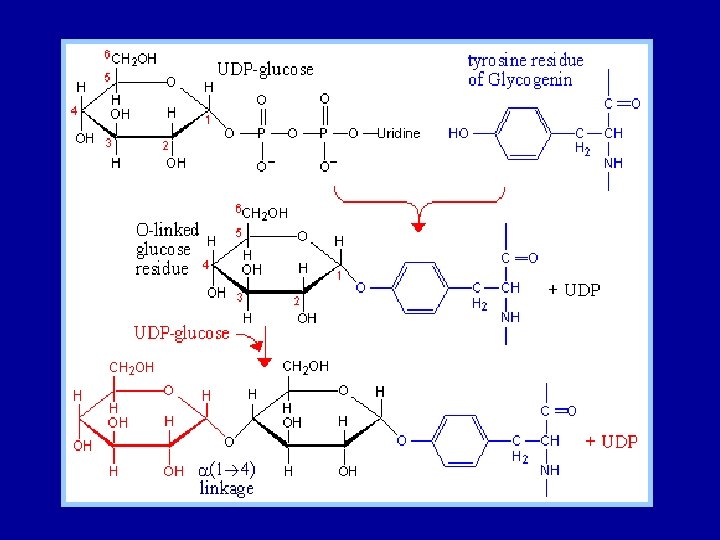
Glycogen is synthesized and degraded by different pathways - Glycogen is synthesized by different pathway from that of glycogen breakdown - UDP-glucose, the activated intermediate in glycogen synthesis, is formed from glucose 1 -phosphate and UDP - Glycogen synthase catalyzes the transfer of glucose from UDP-glucose to the C-4 hydroxyl group of a terminal residue in the growing glycogen molecule - Synthesis is primed by glycogenin, an autoglycosylating protein that contains a covalently attached oligosaccharide unit on a specific tyrosine residue. - A branching enzyme converts some of the alpha-1, 4 linkages into alpha-1, 6 linkages to increase the number of ends so that glycogen can be made and degraded more rapidly.
- In muscle, phosphorylase is activated to generate glucose for use inside the cell as a fuel for contractile activity. - In contrast, liver phosphorylase is activated to liberate glucose for export to other organs, such as skeletal muscle and the brain. - Epinephrine and glucagon stimulate glycogen breakdown through specific 7 TM receptors. - Muscle is the primary target of epinephrine, whereas the liver is responsive to glucagon. Both signal molecules initiate a kinase cascade that lead to the activation of glycogen phosphorylase.
Glycogen breakdown and synthesis are commonly regulated - Glycogen synthesis and degradation are coordinated by several amplifying reaction cascades. - Epinephrine and glucagon stimulate glycogen breakdown and inhibit its synthesis by increasing the cytosolic level of cyclic AMP, which activates protein kinase A. - Elevated cytosolic Ca 2+ levels stimulate glycogen degradation by activating phosphorylase kinase. Therefore, muscle contraction and calcium-mobilizing hormones promote glycogen breakdown. - The glycogen-mobilizing action of PKA are reversed by protein phosphatase 1, that is regulated by several hormones.
- Epinephrine inhibit this phosphatase by blocking its attachment to glycogen molecules and by turning on an inhibitor. - insulin, in contrast, activates this phosphatase by triggering a cascade that phosphorylates that glycogen-targeting subunit of this enzyme. - Hence, glycogen synthesis is decreased by epinephrine and increased by insulin. - Glycogen synthase and phophorylase are also regulated by noncovalent allosteric interactions. - Phosphorylase is a key part of the glucose-sensing system of liver cells. - Glycogen metabolism exemplifies the power and precision of reversible phosphorylation in regulating biological processes.
Glycogen storage diseases 1 - von Gierke’s disease (type l) 2 - Pomp’s disease (type ll) 3 - Cori’s disease (type lll) 4 - Andersen’s disease (type l. V) 5 - Mc Ardle’s disease (type V) 6 - Her’s disease (type Vl) 7 - Tarui’s disease (type Vll) 8 - X-linked phosphorylase kinase deficiency (type Vlll) 9 - Phosphorylase kinase deficiency (type l. X) 10 - Liver glycogen synthase deficiency (type 0)
1 - von Gierke’s disease (type l) G 6 Pase catalyzes the final step leading to the release of glucose into bloodstream by the liver. Deficiency of this enzyme results in an increase of intracellular [G 6 P], which leads to a large accumulation of glycogen of normal structure in the liver and kidney (recall that G 6 P inhibits glycogen phosphorylase and activates glycogen synthase) and an inability to increase blood glucose concentration in response to glucagon or epinephrine. Similar difficulties occur when there are defects in the protein that transports glucose across the liver cell plasma membrane or in any of the proteins that transport glucose, G 6 P, or Pi across the endoplasmic reticulum membrane.
The symptoms of type l glycogen storage disease include: Massive liver enlargement Sever hypoglycemia (low blood sugar) after a few hour fast. General failure to thrive Treatment by drug-induced inhibition of glucose uptake by the liver to increase blood [glucose] Continuous intragastric feeding overnight
2 - Pomp’s disease (type ll) (α-1, 4 -glucosidase deficiency) This is the most devastating glycogen storage disease. It results in a large accumulation of glycogen of normal structure in the lysosomes of all cells and causes death by cardiorespiratory failure, usually before the age 1 year. α -1, 4 -glucosidase functions is to hydrolyze maltose and linear oligosaccharides, as well as the outer branches of glycogen, thereby yielding free glucose.
3 - Cori’s disease (type lll) Amylo-1, 6 -glucosidase (debranching enzyme) deficiency In this disease, glycogen of abnormal structure containing very short outer chains accumulates in both liver and muscle since, in the absence of debranching enzyme, the glycogen cannot be further degraded. Its symptoms is hypoglycemia but not sever as in those of von Gierk’s disease type l. The low blood sugar, that in this case is a result of the decrease efficiency of glycogen breakdown Treated with frequent feeding and a high protein diet.
4 - Andersen’s disease (type l. V) Amylo-(1, 4→ 1, 6)-Transglycosylase (branching enzyme) deficiency This is one of the most sever glycogen storage disease; victims rarely survive past the age 5 years because of liver dysfunction. Glycogen Concentration in liver is not increased but its structure is abnormal, with very long unbranched chains resulting from the lack of branching enzyme. It has been suggested that the liver dysfunction may be caused by a foreign body immune reaction to the abnormal glycogen.
5 - Mc Ardle’s disease (type V) Muscle phosphorylase deficiency Its major symptom, that is most severely obvious in early adulthood, is painful muscle cramps on exertion. This situation is a result of the inability of the glycogen breakdown system to provide sufficient fuel for glycolysis to keep up with the metabolic demand for ATP. 6 - Her’s disease (type Vl) liver phosphorylase deficiency Patients with a deficiency of liver phosphorylase have aymptoms similar to those with mild forms of type l. The hypoglycemia in this case results from the inability of glycogen phosphorylae to respond to the need for glucose production by the liver.
7 - Tarui’s disease (type Vll) muscle phosphofrucktokinase deficiency The result of a deficiency of the glycolytic enzyme PFK-1 in muscle is an abnormal buildup of the glycolytic metabolites G 6 P and F 6 P. Symptoms are similar to those of type V. Since PFK deficiency prevents glycolysis from keeping up with the ATP demand of muscle contraction. 8 - X-linked phosphorylase kinase deficiency (type Vlll) liver phosphorylase kinase deficiency Some individuals with symptoms of type VI glycogen storage disease have liver phosphorylase of normal structure.
However, they have a defective phosphorylase kinase, which results in their inability to convert phosphorylase b to phosphorylase a. 9 - Phosphorylase kinase deficiency (type l. X) Phosphorylase kinase deficiency, when it is autosomal recessive, is caused by a mutation in one of the genes encoding a β, γ, orδ subunit of phosphorylase kinase. Since different organs contain different isozymes of phosphorylase kinase , the symptoms and severity of type IX disease vary with the affected organs.
10 - Liver glycogen synthase deficiency (type 0) This is only disease of glycogen metabolism in which there is a deficiency rather than an accumulation of glycogen. The activity of liver glycogen synthase is extremely low in individuals with this disease and therefore they exhibit hyperglycemia after meals and hypoglycemia at other times. The root cause of type 0 glycogen storage disease is still under investigation.
The metabolism of lipids - Fatty acid β-oxidation – objective - Outline the steps in the β-oxidation and lipogenesis of lipids. - Explain the conditions under which ketone bodies are formed and why - Explain the difference between catabolism and anabolism of lipids - Explain the need for essential fatty acids
Catabolism of lipids – introduction - Fat and glucose are the two major sources of energy for ATP production under most conditions. - Fat provides about half the energy for resting muscle and heart. - The majority of the body’s energy is stored as fat, partly because it is not hydrated and partly because adipose tissue storage appears to be unlimited. - Basically fats converted into acetyl-Co. A and then fed into the citric acid cycle (Krebs).
β-oxidation generates FA and TAG Fat cell glycerol Triacylglycerol pyruvate glucose fatty acids Acetyl-Co. A TCA cycle CO 2 + H 2 O
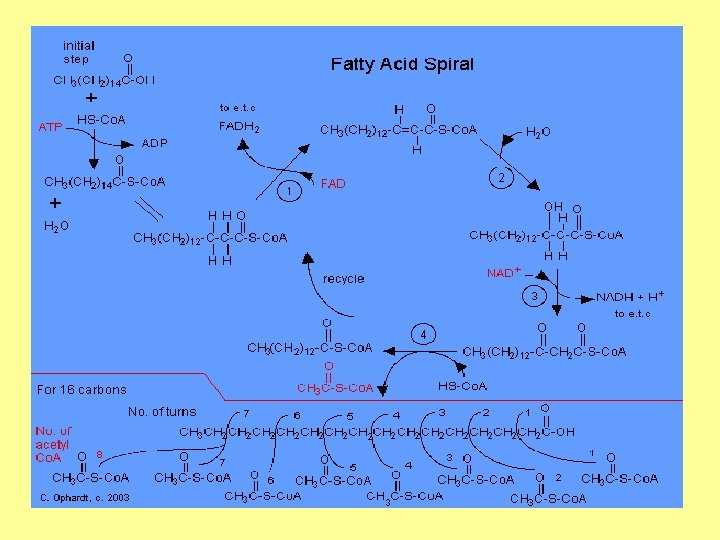
conversion of activated fatty acid to acetyl-Co. A molecules inside the mitochondrion The reactions involved are: 1 - oxidation (an FAD dependent dehydrogenation) 2 - Hydration by enoyl-Co. A hydrates 3 - oxidation (an NAD+ dependent dehydrogenation) 4 - cleavage (Hydrolysis) by thiolase, yielding 1 acetyl-Co. A and a fatty acid that has now been shortened by 2 carbons (acyl-Co. A)
Lipogenesis Anabolism: making lipids Mechanism of fatty acyl-Co. A synthesis 1 - Acetyl-Co. A and malonly-Co. A bond to separate acyl carrier proteins. 2 - Condensation of actyl-ACP and malonyl-ACP to form actoacetyl-ACP. 3 - Reduction by NADPH. 4 - Dehydration. 5 - Reduction by NADPH.
-The cycle now repeats but starting from C 4 rather than C 2, it continues up to C 16. - Aseparate elongation system operates beyond this length. - Steps 2 -4 in lipogenesis are approximate reversals of the corresponding three steps in fatty acid β-oxidation.
Mechanism of fatty acid acyl-Co. A synthesis - The reductant is NADPH rather than NADH - The extra phosphate does not participate in reduction reactions; it is an identification signal, Allows cell to separate reductive power for energy use for synthetic reactions. - NADPH provides reducing power for FA synthesis. - The amount of NADPH generated is only half that which is required for FA synthesis. - The source of the remainder is from the PPP.
Organization of the fatty acid synthesis process - In the bacteria such as E. Coli, there are separate enzymes for each step. - In animals a large enzyme complex called fatty acid synthase carries out all the reactions. The partly synthesized fatty acid remains on the complex until it is complete. - Longer chain fatty acid than palmitate (C 16) are formed by elongation reactions in the endoplasmic reticulum by separate enzymes.
Explain the need for essential fatty acid - Unsaturated fatty acids are required for membrane synthesis and other activities in the body. - Mammals cannot introduce adouble bond beyond C 9 of the FA chain. - Therefore Linoleate (2 double bonds) and Linolenate (3 double bonds) must be included in the diet. - They are essential FA (polyunsaturated) because they are originator of other vital lipids, including prostaglandins.
Summary Fatty acid oxidation - Fatty acid oxidation begins with a dehydrogenation between carbon 2 and 3 of acyl-Co. A derivative. - Next, the trans double bond produced in the first step undergoes enzymatic hydration. - The derivative produced in the second step then undergoes dehydrogenation. There are two final products: 1 - A molecule of acyl-SCo. A from carbons 1 and 2 of the original fatty acid and 2 - Another fatty acyl-SCo. A molecule having 2 fewer carbon atoms than does the original acyl-SCo. A derivative.
The acetyl-SCo. A formed in this process enters the citric acid cycle and is oxidized to CO 2 and water. The Biosynthesis of fatty acid - The biosynthesis of fatty acids begins when citrate leaves the mitochondria and enters the cytosol where the fatty acid synthase complex lies. - Citrate is both the allosteric signal for the initiation of fatty acid biosynthesis and the origin of acetyl-SCo. A, which is the substrate for the first reaction of malonyl. SCo. A. - The central feature of the fatty acid synthase complex is the presence of two unique-SH groups to which growing fatty acid chains are esterified.
- In the first chain-lengthenine step, acetyl-SCo. A is transferred to the OSH group on the condensing enzyme, and malonyl. SCo. A is transferred to the –SH group on the acetyl carrier protein. - The acetyl group is then condensed with the malonyl group to form the acetoacetyl derivate. At the same time, CO 2 is displace and lost. - The acetyl group becomes the methyl terminal group of the growing chain. - In the next three reactions, the –keto group is reduced, dehydrated, and finally reduced to form an aliphatic chain increased by two carbon atoms.
- The process is repeated until a 16 -carbon chain is formed and released by hydrolysis as palmitoyl-SCo. A. Ketone bodies A significant fraction of the acetyl-Co. A produced by fatty acid oxidation in the liver is converted to acetoacetate and D-beta-hydroxybutyrate, which together with acetone are referred to as ketone bodies. The first two compounds serve as important fuels for the peripheral tissues.
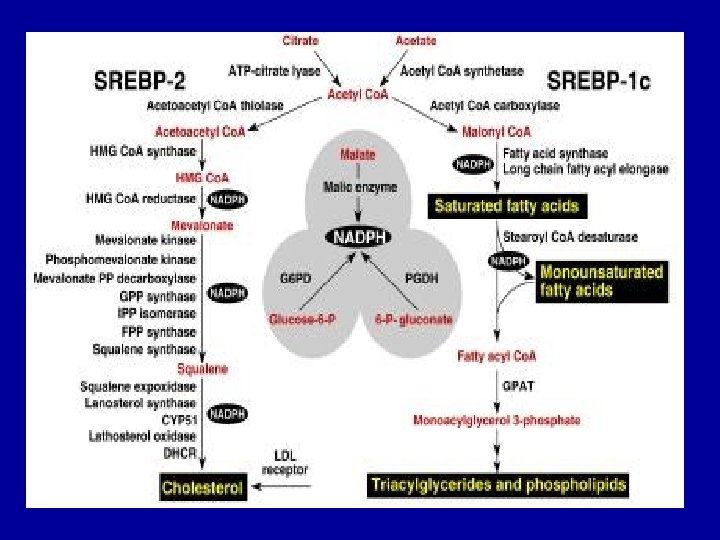
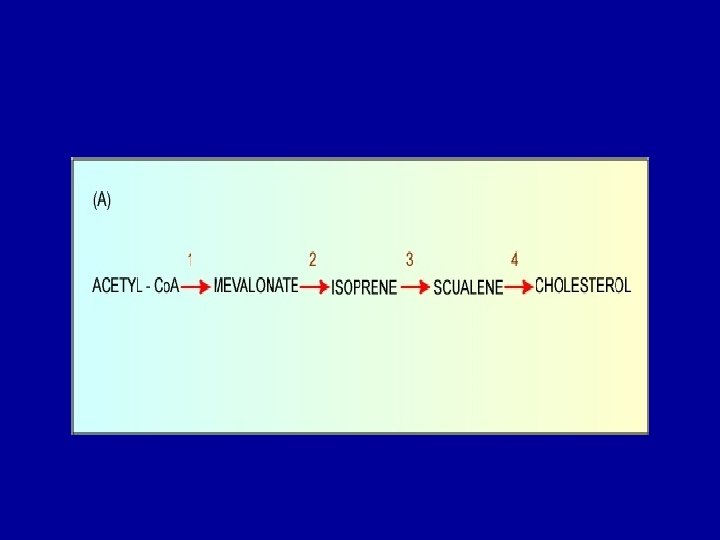
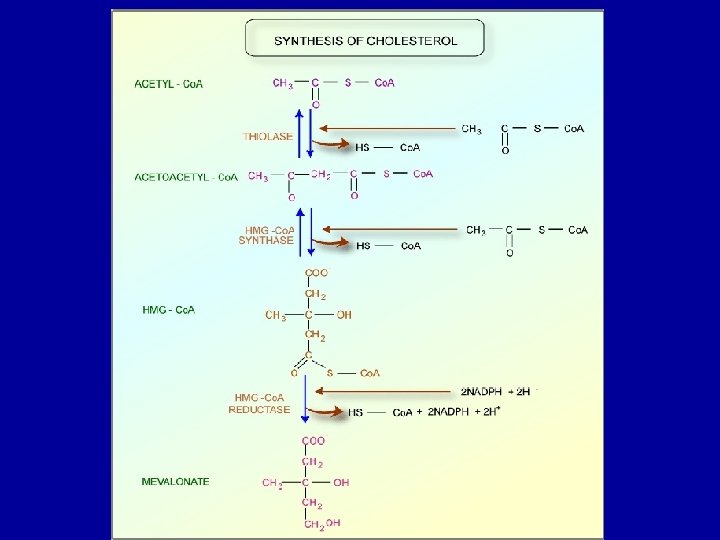
Cholesterol Biosynthesis - Cholesterol is a vital component of cell membranes and is the originator of the steroid hormones and bile salts. - Cholesterol is synthesized in the liver from acetate in a pathway that involves formation of HMG-Co. A from 3 molecules of acetate. -HMG-Co. A followed by reduction, phosphorylation, decarboxylation, and dehydration to form isoprene units isopentenyl pyrophosphate and dimethylallyl pyrophosphate. - 4 of these isoprene units are then condensed via cationic mechanism to form squalene. - Squalene in turn undergoes a cyclization reaction to form lanosterol, the originator to cholesterol.
Points To Remember: - Fatty acid synthesis and degradation are basically the reverse process of each other. - Triacylglycerols are the most common storage and transport form of fatty acids and hence otherwise known as neutral fats. - Fatty acids are synthesized or derived from the diet of an organism. - Liver and adipose tissue are the significant locations for synthesis of most triacylglycerols. - Adipose tissue serves as the primary storage site for triacylglycerols. - A series of more than twenty reactions are involved in the usual metabolic pathway for cholesterol biosynthesis and each reaction is catalyzed by a specific enzyme.
- The liver is the major site of cholesterol biosynthesis. The location of synthesis is cytoplasmic, with acetyl Co. A providing all the necessary carbon atoms. - HMG-Co. A reducatse can be inhibited by drugs of statin class like Lovastatin. - Bile acids and salts serve to emulsify fats that are essential for fat digestion and absorption. - Cholelithiasis is a condition when more cholesterol enters the bile than can be excreted. - The soluble vitamins A, D, E and K are relatives of the basic cholesterol structure.
- The rate-limiting step in the sequence is the synthesis of a six-carbon molecule, mevalonate, catalyzed by the enzyme 3 -hydroxy-3 methylglutaryl Co. A reductase (HMG Co. A reductase). - Hormones are synthesized by adrenal cortex, testis, ovary, and placenta like Adrenal cortical steroid, Aldosterone, Cortisol, Gonadal steroids, Progesterone and Testosterone. These are also structurally related to cholesterol.
Biosynthesis and functions of leukotrienes, prostaglandins and phospholipids Leukotrienes, prostaglandins, thromboxanes, and lipoxins are eicosanoid products produced largely by the metabolism of arachidonate. These highly unstable compounds have profound physiological effects at extremely low concentrations. They are involved in the inflammatory response, the production of pain and fever, the regulation of blood pressure, and many other important physiological processes. Arachidonate is synthesized from linoleic acid, an essential fatty acid, and stored as phosphatidylinositol and other phospholipids.
Prostaglandins and thromboxanes are synthesized via the “cyclic pathway” whereas leukotrienes and lipoxins are synthesized via the “linear pathway. ” Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the cyclic pathway but not the linear pathway. Adipose tissue in starvation Increased synthesis of ketone bodies - Liver is unique in being able to synthesize and release ketone bodies (primarily B-hydroxybutyrate) for use as fuel by peripheral tissues. - Ketone body synthesis is favored when the concentration of acetyl. Co. A that produced from fatty acid metabolism, exceeds the oxidative capacity of the TCA cycle.
- Significant synthesis of ketone bodies starts during the first days of starvation. - The availability of circulating ketone bodies is important in starvation because they can be used for fuel by most of tissues including brain. - Once their level in the blood sufficiently high, this reduces the need for gluconeogenesis from amino acid carbon skeletons, thus slowing the loss of essential protein, - Ketone body synthesized as part of the overall hepatic response to starvation.
Glucose transport into adipocyte and its subsequent metabolism are depressed owing to low levels of circulating insulin, this lead to a decrease in fatty acid and triacylglycerol synthesis. - In starvation, fatty acid obtained from hydrolysis of stored triacylglycerol are released into blood. - When fatty acids bound to albumin, they are transported to a variety of tissues for use as fuel. - The glycerol produced following triacylglycerol degradation is used as a gluconeogenic originator by the liver. - Fatty acids are also converted to acetyl-Co. A, which can enter the TCA cycle, thus producing energy for the adipocyte.
Skeletal muscle in starvation - Glucose transport into skeletal muscle cell via insulin-dependent glucose transport protein in the plasma membrane and subsequent glucose metabolism are depressed because of low levels of circulating insulin. - During the first 2 weeks of starvation, muscle uses fatty acids from adipose tissue and ketone bodies from the liver as fuels. - After about 3 weeks of starvation, muscle decreases its utilization of ketone bodies and oxidizes fatty acids almost exclusively. This leads to a further increase in the already elevated level of circulating ketone bodies
Overview of Diabetes mellitus is a major public health problem affecting 2%-3% of the united states population. It is the leading cause of adult blindness, and a major cause of renal failure, heart attacks, and stroke. Diabetes is not one disease but rather is a diverse group of syndromes characterized by an a relative or absolute deficiency in insulin. There are two groups, insulin-dependent diabetes mellitus (IDDM) and non-insulin-dependent diabetes mellitus (NIDDM), based on their requirements for insulin.
Disorders of Lipid Metabolism - Fats (lipids) are an important source of energy for the body. The body's store of fat is constantly broken down and reassembled to balance the body's energy needs with the food available. - Groups of specific enzymes help the body break down and process fats. Certain abnormalities in these enzymes can lead to the buildup of specific fatty substances that normally would have been broken down by the enzymes. - Over time, accumulations of these substances can be harmful to many organs of the body. Disorders caused by the accumulation of lipids are called lipidoses. Other enzyme abnormalities prevent the body from converting fats into energy normally. These abnormalities are called fatty acid oxidation disorders.
Gaucher's Disease - Gaucher's disease is caused by a buildup of glucocerebrosides in tissues. Children who have the infantile form usually die within a year, but children and adults who develop the disease later in life may survive for many years. - In Gaucher's disease, glucocerebrosides, which are a product of fat metabolism, accumulate in tissues. Gaucher's disease is the most common lipidosis. The disease is most common among Ashkenazi (Eastern European) Jews. Gaucher's disease leads to an enlarged liver and spleen and a brownish pigmentation of the skin. Accumulations of glucocerebrosides in the eyes cause yellow spots called pingueculae to appear. Accumulations in the bone marrow can cause pain and destroy bone.
Sphingoglcolipid degradation and lipid storage diseases Sphingoglcolipids are lysosomally degraded by a series of enzymatically mediated hydrolytic reactions. These reactions are catalyzed at the lipid-water interface by soluble enzymes, often with the aid of sphingolipid activator proteins (SAPs). These nonenzymmatic ancillary proteins are thought to increase the convenience of the carbohydrate moiety of the sphingolipid to the degradation enzyme. The hereditary absence of one of the sphingolipid hydrolases or SAP results in a sphingolipid storage disease. The most common condition is Tay-sach disease, an autosomal recessive deficiency in hexosaminidase A, that results in the neuronal accumulation of GM 2.
Although infants born with Tay-Sachs disease at first appear normal, by ~1 year of age, when sufficient GM 2 has accumulated to interfere with neuronal function, they become progressively weaker, retarded, and blinded until they die, usually by the age of 3 years.
Lipoproteins Definition: lipids and proteins associate noncovalently to form lipoproteins, which function in the blood plasma as transport vehicles for triacylglycerols and cholesterol. Classes of lipoprotein: Lippproteins have been classified into five categories on the basis of their functional and physical properties. 1 - Chylomicrons: which transport exogenous (externally supplied, e. g, dietary) triacylglycerols and cholesterol from the intestines to the tissues.
2 -4 Very low density lipoproteins (VLDL), intermediate density lipoproteins (IDL), and low density lipoproteins (LDL): A group of related particles that transport endogenous (internally produced) triacylglycerols and cholesterol from the liver to the tissues (the liver synthesize triacylglycerols from excess carbohydrates). 5 - High density lipoproteins (HDL): which transport endogenous cholesterol from the tissues to the liver.
Lipoprotein Dysfunction in Atherosclerosis and Alzheimer’s disease Atherosclerosis, the most common form of arteriosclerosis (hardening of the arteries), is characterized by the presence of artheromas, arterial thickening that on sectioning, show a pale yellow deposit of almost pure cholesteryl esters. a) Deficient LDL receptors result in Atheroclerosis. The development of atherosclerosis is strongly correlated with the level of plasma cholesterol. This is particularly evident in individuals with familial hypercholesterolemia (FH). Homozygote with this inherited disorder have such high levels of the cholesterol-rich LDL in their plasma that their plasma cholesterol levels are 3 -5 fold greater than the average level of ~175 mg/100 ml.
However, fat greater damage is caused by the rapid formation of atheromas that, in homozygotes, cause death from myocardial infraction as early as the age of 5. Heterozgotes, which comprise ~1 person in 500, are less severely cause problems; they develop symptoms of coronary artery diseae after the age 30. Cells taken from FH homozygotes completely lack functional LDLR, whereas those taken from heterozygotes have about half of the normal balance. Homozygotes and to a less extent, heterozygotes are therefore unable to utilize the cholesterol in LDL. Rather, their cells must synthesize most of the cholesterol for their needs. The high level of plasma LDL in these indivituals results from 2 causes:
1 - its decreased rate of degradation because of the lack of LDLR 2 - its increased rate of synthrsis from IDL due to the failure of LDLR to take up IDL. b) Scavenger receptors take up oxidized LDL c) Atherosclerosis is a multifactorial disease d) Apo E 4 is implicated in both cardiovascular disease and Alzheimer’s disease e) Apo E 2 has a low affinity for LDL receptor