CONDUITE A TENIR DEVANT UNE CHOLESTASE DR REZAK








. • Entéropathie")



 Confirmer la Cholestase. 2)Evaluer la gravite : - Urgence. -")







 : anomalie biliaire est")

















- Slides: 40
CONDUITE A TENIR DEVANT UNE CHOLESTASE DR REZAK R
INTRODUCTION DEFINITION : Clinique : selles décolorées + urines foncées. Physiopathologie : ensemble des manifestations dues : - à la diminution Ductules, canaux ou biliaires et VB : modification de la bile -à l’arrêt du flux biliaire ou - à une anomalie de formation de la bile. (Pôle biliaire de l ’hépatocyte = B. primaire ou hépatique Sécrétion de : Bili C, cholestérol, acides biliaires, HCO 3 - …)
ANATOMIE DU FOIE
PHYSIOPATHOLOGIE
PHYSIOPATHOLOGIE: Mécanisme
CONSEQUENCES • Absence des sels biliaires dans la lumière intestinale: Malabsorption des graisses (TCL, AGE). Malabsorption des Vitamines liposolubles (ADEK). • Rétention des acides biliaires : Prurit. Lésions hépatocellulaires. FIBROSE , CIRRHOSE HTP IHC • Conséquences d’obstacle extra hépatique : • Cholangite bactérienne. • Pancréatite aigue : – Sténose de la VBP –Rupture de la VBP • Péritonite biliaire
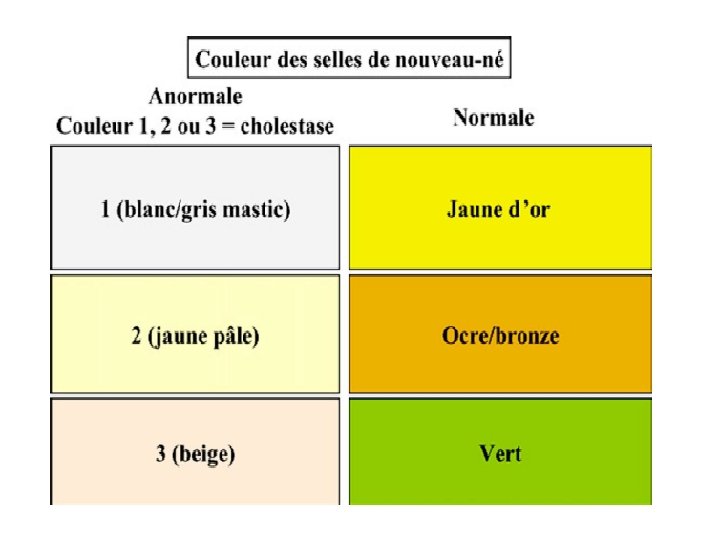
CLINIQUE • COULEUR DES SELLES TYPE DE DECOLORATION : Complete et totale : nouveau ne : AVBEH ± MUCOVISCIDOSE , deficit en Alpha 1 Incomplète et intermittente : Maladies métaboliques , Génétiques.
Clinique • Hépatomégalie : a recher systématiquement • Dure et foie median : AVBEP • Splénomégalie : peut s y associer ( signes HTTP , penser a des maladies métaboliques ou de surcharges : Niemann pick, Gaucher)
Clinique • Douleur abdominal, Vomissements. • Prurit ( accumulation des acides biliaires). • Entéropathie Exsudative. • Malnutrition par malabsorption. • Rachitisme par malabsorption. • ………
Clinique : Signes de Gravites Signes d’HTP clinique : - Splénomégalie. - Signes d’hypersplenismes. - Circulation collatérale. -Ascite. -Hémorragies d’HTP : hématémèse par rupture des varices oesophagiennes. rectorragies par ruptures des veines rectales.
Clinique : Signes de Gravite -Cirrhose et Encéphalopathie Hépatique : Classification des EH : sta des Niveau de conscience Personnalité et intellect Signes neurologiques Anomalies EEG I Inversion du rythme du sommeil fatigue Trouble de la concentration. Confusion légère. Irritabilité. Troubles de la coordination. Apraxie. Finger tremor (trouble de l’ecriture). Activite polyrythmique(5 HZ) Réactivité EEG II léthargie Désorientation. Amnésie. Flapping tremor. Hyporeflexie. Dysarthrie. Activité lente théta-delta prédominante(3 -4 Hz). Réactivité inconstante. III Somnolence. Confusion. Désorientation. Agressivité. Flapping tremor. Hyporeflexie. Signe Babinski. Rigidité musculaire Activité lente delta monomorphe(1 -3 Hz). Pas de réactivité. IV Coma. Aucun. Décérébration. Convulsions. Tracé déprimé (˂1 Hz) Amplitude décroissante. Pas de réactivité. V EEG quasi plat ou plat.
Clinique : Signes de Gravite Les signes a craindre et a recher devant les obstacles extra-hépatiques : • Cholangite bactérienne: douleur + fièvre. • Pancréatite aigue : – Sténose de la VBP. –Rupture de la VBP. • Péritonite biliaire.
QUELS BILANS REALISES 1) Confirmer la Cholestase. 2)Evaluer la gravite : - Urgence. - Complication. 3) Bilans Etiologiques
Confirmer la cholestase 1/ Le bilan hépatique - Bilirubines totale et conjuguée Ictère à BC = BC ˃ 20% DE BT - Transaminases (ASAT-ALAT) : Marqueur de cytolyse hépatique (souffrance hépatocytaire) ASAT : également marqueur musculaire… - Phosphatases alcalines (PAL) : Elevées cholestase intra ou extrahépatique Attention : également en cas de certaines affections osseuses - Gamma-glutamyl transpeptidase (gamma-GT) Test sensible d’altération hépatique mais peu spécifique.
Confirmer la cholestase 2/bilan d’hemostase : Facteurs de la coagulation : Synthétisés par le foie - I, II, V, VII, IX, X sont vit-K dépendants - I, II, V, VII, IX, X sont explorés par le Temps de Quick 3/ Albuminémie - Exclusivement synthétisée par le foie - Reflet de l’insuffisance hépatocellulaire - Attention : penser aux autres causes d’hypoalbuminémie fuite urinaire : syndrome néphrotique fuite digestive : entéropathies exsudatives
Confirmer la cholestase ECHOGRAPHIE ABDOMINALE : Foie : taille et contours échostructure homogénéité? voies biliaires vésicule Doppler : veines sus-hépatiques veine porte
Biopsie Hépatique • Le recours a la biopsie n est pas systématique. • Ses indications sont bien précises : Bilan étiologique négatif
ETIOLOGIES : Selon Mécanisme ATTEINTE HEPATOCYTE ATTEINTE CANALICULAIRE ATTEINTE VOIE BILIAIRE ¤ Déficit synthèse ¤ Atrophie (paucité). ¤ Atrésie. ¤ Obstruction (sténose). ¤ Obstruction ¤ Déficit transport ¤ Déficit sécrétion.
ETIOLOGIES CAUSES INTRAHEPATIQUES CAUSES EXTRAHEPATIQUE Causes Génétiques Causes Non Génétiques Lithiase biliaire Déficit de synthèse des acides biliaires embryofoetopathie Kyste du cholédoque Syndrome d’Alagille infection Cholestase fibrinogenes familiales toxique Déficit en alpha 1 antitrypsine Déficit en cortisol Sténose congénitale Maladies métaboliques Atrésie des VB Cholangite sclérosante Mucoviscidose Perforation des VB CAUSES EXTRA ET INTRAHEPATIQUE S transitoire
Atrésie Des Voies Biliaires L’atrésie des voies biliaires est responsable d’environ la moitié des Cholestases néonatales. C’est une maladie congénitale mais non héréditaire, de tout l’arbre biliaire, de cause inconnue, commençant aux alentours de la naissance et s’étendant de l’extérieur vers l’intérieur du foie, entraînant une occlusion complète de la lumière du canal biliaire. La conséquence en est une cirrhose biliaire, par les lésions directes des canaux, et la rétention de bile.
AVB : Clinique AVB NON SYNDROMIQUE AVB non syndromique (90%) : anomalie biliaire est isolée. AVB SYNDROMIQUE AVB syndromique (10%- 25%) Malformations congénitales digestives et cardiaques associées: Echographie ++ SYNDROME DE POLYSPLENIE : Prédominance féminine Anomalies digestives composants de ce syndrome : - une polysplénie, ou asplénie ou double rate. - une absence de veine cave inférieure. - une veine porte préduodénale. - une anomalie de la vascularisation artérielle hépatique - un situs inversus - une malrotation digestive - un poumon droit bilobé ou levo-isomérisme pulmonaire en cas d'asplénie. (1) Anomalies cardiaques associées : 30% des cas CIA, CIV défaut septal sténose pulmonaire, coartion de l'aorte hypoplasie du V gauche. (2) 1)Chandra R. Biliary atresia and other structural anomalies in the congenital polysplenia syndrome. J Pediatr 1974; 85(5): 649 -655 2)Davenport M Biliary atresia splenic malformation syndrome : an etiologic and prognostic subgroup. Surgery 1993; 113(6): 662 -668
AVB : Aspects Radiologiques : jeun d au moins 12 heures+++++ - Dans 20% des cas l’échographie est d’aspect normal
AVB : Aspects Radiologiques : jeun d au moins 12 heures+++++
Classification des AVB C Chardot. Epidemiology and management of biliary atresia in french: result 1986 -2009.
Traitement Chirurgical :
Atrésie des voies biliaire: survie et pronostic C Chardot. Epidemiology and management of biliary atresia in french: result 1986 -2009.
Paucité des voies biliaires Syndrome d ’Alagille : 3/5 critères majeurs : -Cholestase avec hypercholestérolémie et hypoplasie ductuaire -RP -Faciès : Front bombé, petit menton, ensellure nasale, hypertélorisme -Embryotoxon. -Vertèbres en aile de papillon. Critères mineurs : -Retard de croissance et/ou mental et/ou pubertaire -Atteinte rénale : tubulaire et/ou glomérulaire (mésengiolipidose)
Cholestases Fibrinogènes Familiales Maladies autosomiques récessives. 03 types : PFIC 1, 2, 3 (Byler’s disease). GGT normal PFIC 1(FIC 1) PFIC 2 (BSEP) augmente PFIC 3 (MDR)
Cholestases Fibrinogènes Familiales PFIC 1 PFIC 2 -Prurit. -GGT et cholestérol normaux -Prolifération ductulaire, bouchons , fibrose, cirrhose. -Arbre biliaire normal. -Baisse de la concentration des acides biliaires. -Lithiase. -Apparition tardive. -Diarrhée. -Période néonatale. -↗↗↗alpha foeto et transaminases. -Lithiase. -Carcinome. PFIC 3 -Période néonatale jusqu’au jeune adulte. -Prurit modéré. -Prolifération ductulaire, fibrose portale, cirrhose. -Arbre biliaire normale. -↗rapport acides biliaires/phospholipides. -Lithiase. -hypertension portale et cirrhose.
Cholestases Fibrinogènes Familiales
Déficit de synthèse des acides biliaires Il est reconnu actuellement que la cholestase et lesions hépatiques peuvent resulter du deficit du synthese de l’acide biliaire primitif, acide cholique et l’acide chenodeoxycholique(CDCA). Ces anomalies representent environ 2% des syndromes cholestatiques de l’enfant. Mais leur evolution a long terme et pronostic bon a long terme , nous obligent a les connaitres.
Déficit de synthèse des acides biliaires Joel M. Andres, MD and Allah B. Haafiz. Neonatal Cholestasis
INFECTIONS • Chez le nouveau né et le nourrisson: -Infections bactériennes type : infection urinaire à Ecoli -Embryofoetopathies. • Chez l’enfant et l’adolescent: -Hépatite virale : Hépatite A+++++++.
TRAITEMENT • Le traitement est étiologique. • Souvent dans les formes avancées , le traitement est symptomatique.
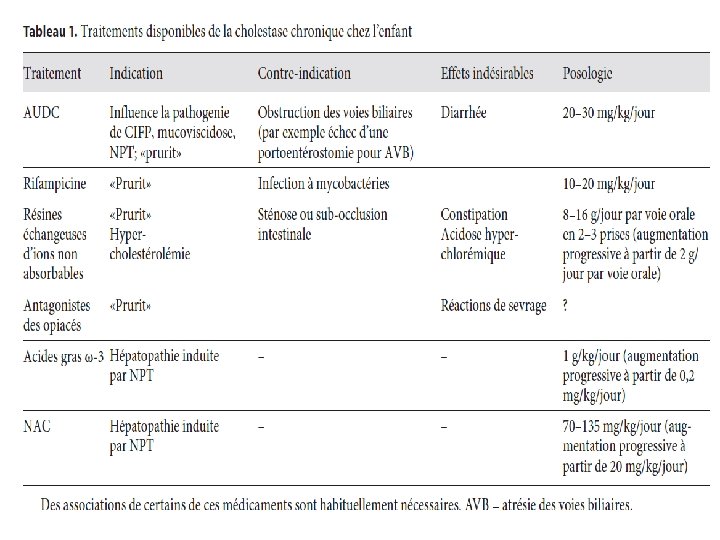
TRAITEMET SYMPTOMATIQUE Devant toute cholestase chr • Vitaminothérapie : Vitamine K : 10 mg/15 J en IVL. Vitamine D: 200000 UI/3 mois. IM profonde. Vitamine E: 15 mg/kg/15 J (max 200 mg). IM profonde. Vitamine A: 150000 UI/2 mois. IM profonde. • Acide ursodesoxycholique : AUDC acide biliaire naturelle , utilisée chez l’enfant dans : -Atresie des Voies Biliaires. -Syndrome d’Alagille. -Cholestases familiales fibrinogenes. -Déficit cong de synthese des acides biliaires. -Mucoviscidose. - cholestase associée a l’alimentation parenterale. Posologie : 600 mg/m 2/J EN 2 prises (20 à 30 mg/kg/j).
TRAITEMET SYMPTOMATIQUE Devant toute cholestase chr • Albumine et lasilix : en cas de décompensation • Rifampicine : 10à 15 mg/kg
ALIMENTATION • Alimentation hypercalorique : 170 -200%. • Hyperprotidique : 3 g/kg/j. (sauf si IHC). • Hyperglucidique : 24 g/kg/j. • Hyperlipidique: 6 g/kg/j+++TCM˃50%(70%). • Volume: 150 -200 ml/kg/j( retention hydro-sodée).
CONCLUSION : messages à retenir Ne pas s’inquiéter, à tort, de la couleur blanche des selles : la précocité de la décoloration des selles est un argument très important en faveur du diagnostic d’atrésie des voies biliaires. Tout ictère se prolonge au-delà du 10 e jour n’est pas physiologique Porter par excès un diagnostic d’ictère au lait de mère Etre rassuré à tort par une courbe de croissance correcte : la croissance des enfants atteints d’atrésie des voies biliaires est le plus souvent normale pendant les premières semaines Fausser l’interprétation de la couleur des selles par un régime ou un traitement inappropriés (laits synthétiques : couleur grise des selles ; fer: couleur grise ; amphotéricine orale : couleur orange) ; Etre rassuré à tort par à un compte-rendu échographique décrivant une vésicule biliaire présente : la présence d’une vésicule biliaire n’exclut pas le diagnostic d’atrésie des voies biliaires limitée aux canaux hépatiques.