Structure and Function Suggested readings Lee D Redfern












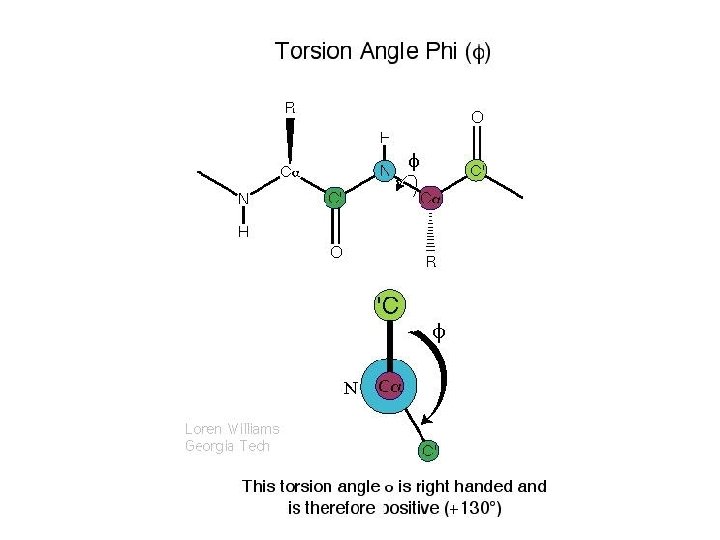
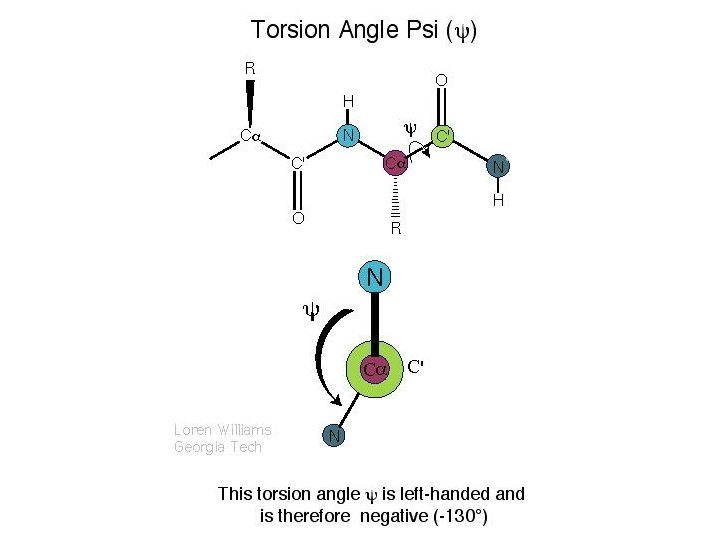
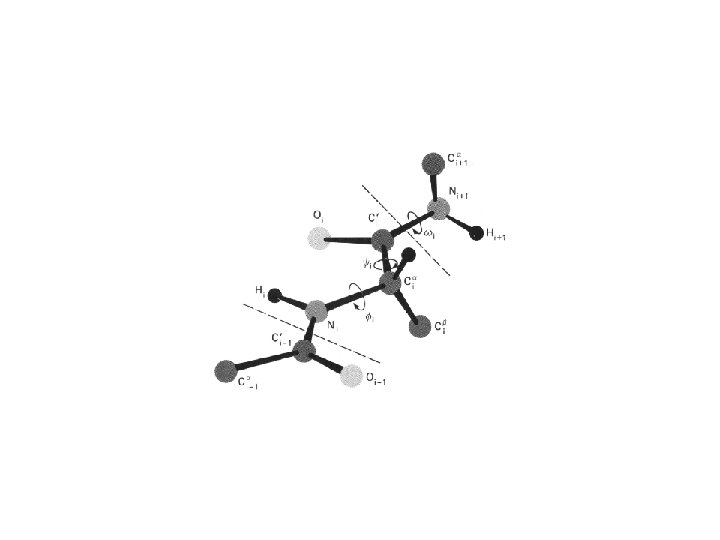
, is")

 in which every backbone N-H group donates a hydrogen")
















 is a repository for 3 -D structural data of")


")
















 to describe gene products in terms of")


- Slides: 63
Structure and Function Suggested readings: Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007 8(12): 995 -1005. Chapter 2 of the book Understanding bioinformatics Marketa Zvelebil Jeremy O. Baum Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O'Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (Uni. Prot): an expanding universe of protein information. Nucleic Acids Res. 2006 Jan 1; 34(Database issue): D 187 -91. Kinoshita K, Nakamura H. Protein informatics towards function identification. Curr Opin Struct Biol. 2003 Jun; 13(3): 396 -400. Review.
The information on biological data is not stored in books…. . BUT in Electronic Databases publicly available (generally!) and accessible by internet Most of the work of a bioinformatician is to learn how to retrieve those data, how to analyse them, and how to produce novel data necessary to the understanding of the complex biological world “Empirical art”
In 1965 Gordon Moore, co-founder of Intel noticed that: Every chip had a capacity double to the predecessor and that every 18 -24 months a new generation of chip was born.
In the seventies Dickerson, a professor of Physical Chemistry noticed that the number of solved X-ray structures of proteins had increased from 1 in 1961 to 23 in 1977
After the completion of the human genome project we have available millions of sequences which represent a biological ‘knowledge’ that we have to understand…
Growth of pdb structures Structures are collected in the PDB databank: http: //www. rcsb. org/pdb/home. do 45. 000 So our ‘knowledge’ is very limited
Protein Structure Prediction • In theory, a protein structure can be solved computationally • A protein folds into a 3 D structure to minimizes its free potential energy • The problem can be formulated as a search problem for minimum energy • • the search space is enormous the number of local minima increases exponentially Computationally it is an exceedingly difficult problem
protein prediction. vs. protein determination X-Ray Threading inferred data Comparative Modeling Experimental data NMR Ab-initio 8
…. from a string of pearl to a folded structure….
Why is it useful to know the structure of a protein, not only its sequence? • The biochemical function (activity) of a protein is defined by its interactions with other molecules. • The biological function is in large part a consequence of these interactions. • The 3 D structure is more informative than sequence because interactions are determined by residues that are close in space but are frequently distant in sequence. In addition, since evolution tends to conserve function and function depends more directly on structure than on sequence, structure is more conserved in evolution than sequence. The net result is that patterns in space are frequently more recognizable than patterns in sequence. 11
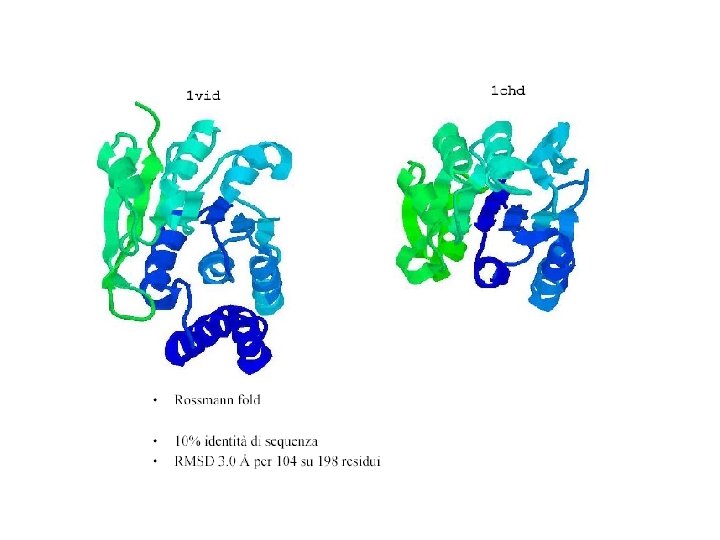
There is a relationship between sequence similarity and structural similarity • Seq. Id. > 50%: core region ~90% of the structure, r. m. s. d. of the main chain around 1. 0 Å • Seq. Id. < 20%: core region ~50% of the structure, r. m. s. d. of main chain around 1. 8 Å [Chothia & Lesk, EMBO J. (1986) 5: 823 -826]
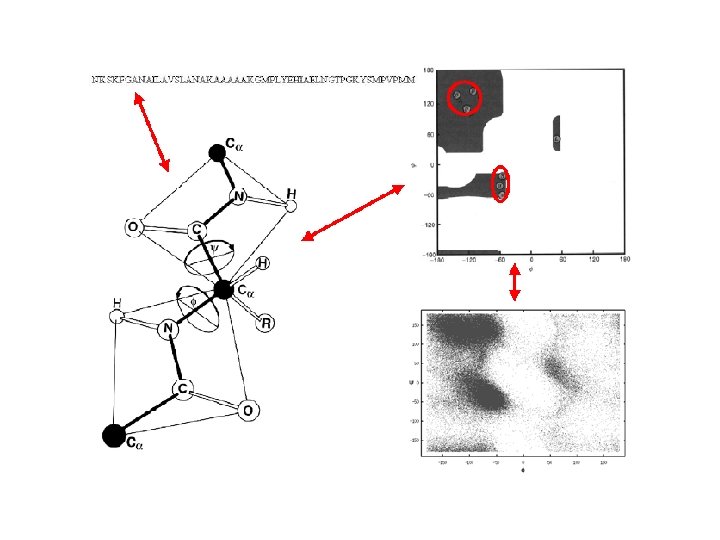
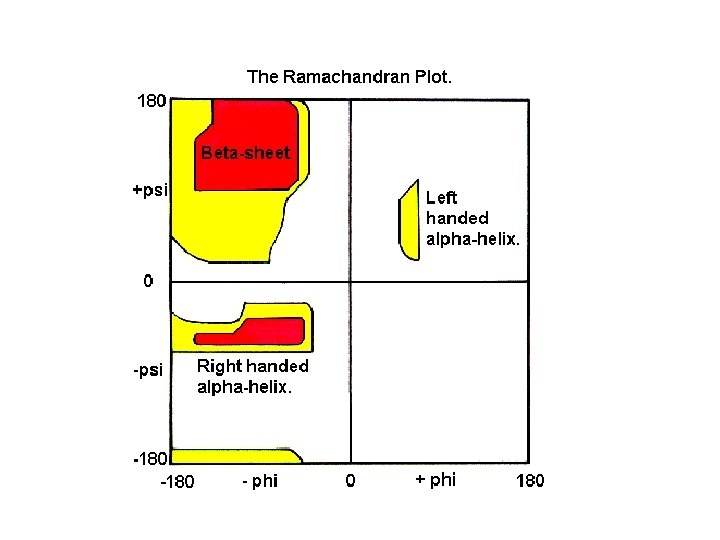
A Ramachandran plot (also known as a Ramachandran map or a Ramachandran diagram), is a way to visualize dihedral angles φ against ψ of amino acid residues in protein structure. It shows the possible conformations of φ and ψ angles for a polypeptide. Mathematically, the Ramachandran plot is the visualization of a function (torus). Hence, the conventional Ramachandran plot is a projection of the torus on the plane, resulting in a distorted view and the presence of discontinuities. One would expect that larger side chains would result in more restrictions and consequently a smaller allowable region in the Ramachandran plot. In practice this does not appear to be the case; only the methylene group at the β position has an influence. Glycine has a hydrogen atom, with a smaller van der Waals radius, instead of a methyl group at the β position. Hence it is least restricted and this is apparent in the Ramachandran plot for Glycine for which the allowable area is considerably larger. In contrast, the Ramachandran plot for proline shows only a very limited number of possible combinations of ψ and φ. A Ramachandran plot generated from the protein PCNA, a human DNA clamp protein that is composed of both beta sheets and alpha helices (PDB ID 1 AXC). Points that lie on the axes indicate N- and C-terminal residues for each subunit. The green regions show possible angle formations that include Glycine, while the blue areas are formations that don't include Glycine. From Wikipedia
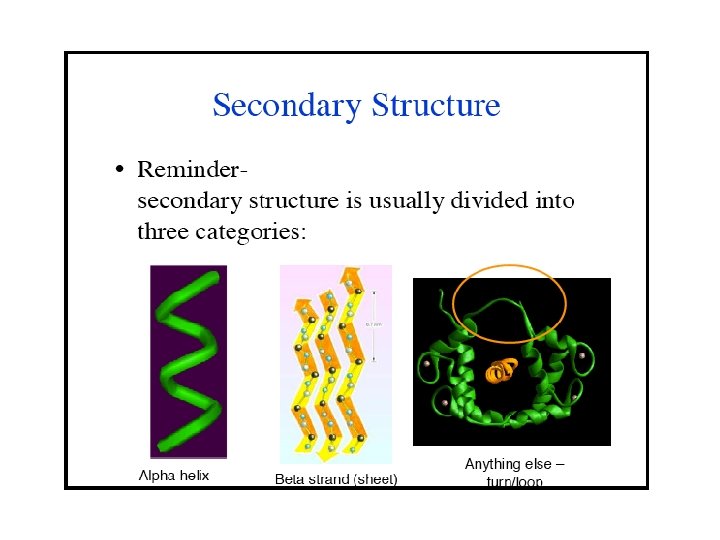
http: //swift. cmbi. ru. nl/gv/dssp/ The DSSP code http: //bioweb. pasteur. fr/seqanal/interfaces/dssp-simple. html H = alpha helix B = residue in isolated beta-bridge E = extended strand, participates in beta ladder G = 3 -helix (3/10 helix) I = 5 helix (pi helix) T = hydrogen bonded turn S = bend Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983 Dec; 22(12): 2577 -637. The secondary structure assignment with DSSP over a database of structures can be used as ‘standard of truth’ for secondary structure prediction methods.
the alpha helix (α-helix) in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier ( hydrogen bonding). The amino acids in a 310 -helix are arranged in a right-handed helical structure. The N-H group of an amino acid forms a hydrogen bond with the C = O group of the amino acid three residues earlier; this repeated i + 3 → i hydrogen bonding defines a 310 -helix.
Homology: two proteins are homologous if they have a common ancestor. This is a binary property (once defined is eithere or not). It is a useful information: when a known gene G is homologous to an unknown gene X we will gain information on X from G through transitivity.
Orthology Homologous sequences are orthologous if they were separated by a speciation event: when a species diverges into two separate species, the divergent copies of a single gene in the resulting species are said to be orthologous. Orthologs, or orthologous genes, are genes in different species, that are similar to each other because they originated from a common ancestor.
Paralogy Homologous sequences are paralogous if they were separated by a gene duplication event: if a gene in an organism is duplicated to occupy two different positions in the same genome, then the two copies are paralogous. A set of sequences that are paralogous are called paralogs of each other. Paralogs typically have the same or similar function, but sometimes do not: due to lack of the original selective pressure upon one of copy of the duplicated gene, this copy is free to mutate and acquire new functions.
Homologies: Rouse Hemoglobin α Hemoglobin Rouse Mouse α Hb Gene duplication β Hemoglobin Speciation Rat α Hb Paralogs Rouse Mouse β Hb Rat β Hb Orthologs
The genes encoding myoglobin and hemoglobin are considered to be ancient paralogs. Similarly, the four known classes of hemoglobins (hemoglobin A, hemoglobin A 2, hemoglobin S, and hemoglobin F) are paralogs of each other. While each of these genes serve the same basic function of oxygen transport, they have already diverged slightly in function: fetal hemoglobin (hemoglobin F) has a higher affinity to oxygen than adult hemoglobin. Paralogous genes often belong to the same species, but this is not necessary: for example, the hemoglobin gene of humans and the myoglobin gene of chimpanzees are paralogs. This is a common problem in bioinformatics: when genomes of different species have been sequenced and homologous genes have been found, one can not immediately conclude that these genes have the same or similar function, as they could be paralogs whose function has diverged.
Where it all starts…. . http: //www. expasy. ch/sprot/
- sequence + conservation structure function Function is attributed to very few atoms absolutely conserved during the evolutionary process
The ‘centrality’ of a 3 D structure
How Can We Compare Sequences ? The Twilight Zone %Sequence Identity Similar Sequence Similar Structure Different Sequence Structure ? ? Same 3 D Fold 30% 30 Twilight Zone Length 100
How Do Sequences Evolve ? In a structure, each Amino Acid plays a Special Role -+ On the surface, CHARGE MATTERS In the core, SIZE MATTERS
Divergent evolution?
Convergent evolution? Inactivation of neurotransmitters Sensorial cellular response
sequence identity for 104 over 198 residues
Structures are collected in the PDB databank http: //www. rcsb. org/pdb/home. do
The Protein Data Bank (PDB) is a repository for 3 -D structural data of proteins and nucleic acids. This data, typically obtained by X-ray crystallography or NMR spectroscopy, is submitted by biologists and biochemists from around the world, is released into the public domain, and can be accessed for free. The database is the central repository for biological structural data. PDB COORDINATE FILE FORMAT Figure 1: Each separate column in a given section of a PDB file is designated as a different "field”
#Definition of PDB format: #http: //www. wwpdb. org/documentation/format 23/v 2. 3. html #The ATOM record: # #COLUMNS DATA TYPE FIELD DEFINITION #----------------------------------------# 1 - 6 Record name "ATOM " # 7 - 11 Integer serial Atom serial number. #13 - 16 Atom name. #17 Character alt. Loc Alternate location indicator. #18 - 20 Residue name res. Name Residue name. #22 Character chain. ID Chain identifier. #23 - 26 Integer res. Seq Residue sequence number. #27 AChar i. Code for insertion of residues. #31 - 38 Real(8. 3) x Orthogonal coordinates for X # in Angstroms. #39 - 46 Real(8. 3) y Orthogonal coordinates for Y # in Angstroms. #47 - 54 Real(8. 3) z Orthogonal coordinates for Z # in Angstroms. #55 - 60 Real(6. 2) occupancy Occupancy. #61 - 66 Real(6. 2) temp. Factor Temperature factor. #73 - 76 LString(4) seg. ID Segment identifier, # left-justified. #77 - 78 LString(2) element Element symbol, # right-justified. #79 - 80 LString(2) charge Charge on the atom.
Atomic coordinates
Contact plots Calculate the distances between C-beta atoms Define a distance cut-off (7 Angstrom) If distance is shorter than cut-off define it as a contact $atom. Dist = sqrt(($x. Coor[$c. Beta[$i]] - $x. Coor[$c. Beta[$j]])**2 + ($y. Coor[$c. Beta[$i]] - $y. Coor[$c. Beta[$j]])**2 + ($z. Coor[$c. Beta[$i]] - $z. Coor[$c. Beta[$j]])**2);
Human prion protein Ovine prion protein variant R 168
Ovine prion Sheep prion
Divergent evolution?
Divergent evolution?
Convergent evolution? Inactivation of neurotransmitters Sensorial cellular response
Convergent evolution?
Proteins are organised in domains with specific architecture So proteins with similar function have similar domain architecture!!
Domains: conserved folded units stable in isolation DTGM Fusion Protein to Treat Patients With Recurrent or Refractory Acute Myeloid Leukemia Anticancer drug formed by the combination of diphtheria toxin and a colony- stimulating factor (GM-CSF)
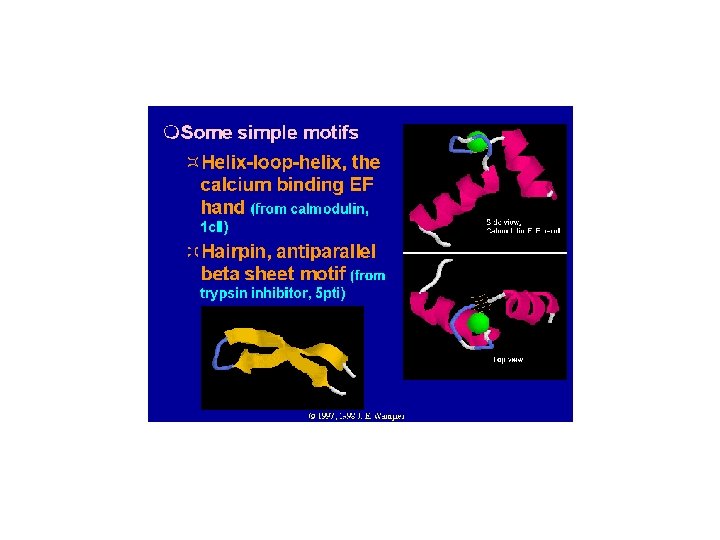
Calmodulin
http: //structbio. vanderbilt. edu/cabp_database/pic_gallery/ca_coord/cam_chapter 1. html
EF-hand Sequences in Pfam Database 1 3 5 7 9 12 X Y Z # -X -Z
Protein function can be divided into three broad areas: molecular function, biological process and cellular component. Molecular function describes activity at the molecular level, such as catalysis, which is commonly predicted through methods that identify homologues or orthologues. Biological process describes broader functions that are carried out by assemblies of molecular functions, such as a particular metabolic pathway. Genomic inference methods can identify the direct physical protein–protein interactions and indirect functional associations that are found in biological processes. Cellular component describes the compartment(s) of a cell in which the protein performs its function. This component can be predicted through methods that predict signal sequences, residue composition, membrane association or post-translational modifications. Within these areas there are broad categories of computational methods, all of which ultimately depend on experimental data. From Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007 8(12): 995 -1005
What does it means function? EC nomenclature
Go project provides three structured vocabularies (ontologies) to describe gene products in terms of their 1)associated biological processes; 2)cellular components; 3)molecular functions http: //www. blast 2 go. de/ http: //www. geneontology. org/GO. annotation. shtml
From Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007 8(12): 995 -1005
Learning outcomes of the lecture The relevance of the knowledge of a 3 D structure: why? Implications? What are the most common secondary structure elements What are the ‘teachings’ of the PDB database How can we approach the search of the function of a protein or a gene