Implementering af den kommende EU Clinical Trial forordning
 Lene Grejs Petersen, Chefkonsulent, Sektion")

 levede ikke op")




 Cover letter EU application form Protocol Investigator’s Brochure IMPD GMP compliance")








 er i gang")

 Kapacitit: Ø 18 nye ansøgninger hver uge (stor variation) Ø Work")


 Ø National Contact Point – Lægemiddelstyrelsen (LMST) Ø Faciliterer procedurerne")
 ‒")
 kan sponsor vælge • At bruge EU-portalen")



- Slides: 28
Implementering af den kommende EU Clinical Trial forordning (536/2014) Lene Grejs Petersen, Chefkonsulent, Sektion for kliniske forsøg
Implementering af forordningen ‒ Lidt historie ‒ Forordning om kliniske forsøg ‒ Hvad gør vi DK? ‒ Overgangsordning ‒ Forberedelse til forordningen 2 27. OKTOBER 2020
Historien om den nye forordning ‒ Direktiv 2001/20/EC (implementeret i 2004) levede ikke op til forventninger – Fald i antal kliniske forsøg, stigning i omkostning, stadig store nationale forskelle ‒ 2008: Indleder proces til udskiftning af direktiv ‒ 2012: Forslag til forordning ‒ 2014: Forordning indføres – implementeres ultimo 2018 (et halvt år efter man har godkendt en EU portal og database, som er en central del af forordningen) ‒ Direktiv: Bindende krav, der implementeres i national lovgivning ‒ Forordning: Gældende ved lov i alle EU lande, uden implementering i national lovgivning
Forordning 536/2014 trådte i kraft 27 Maj 2014, men bliver først anvendt 6 måneder efter at der har været en positiv audit af den europæiske database og portal
Nye regulatoriske krav til kliniske forsøg i Europa Forordningen skal biddrage til at øge antallet af forsøg i Europa (stimulere innovation og forbedre patientbehandling) Ø One stop shop (EU-portal and database) – harmoniserede krav Ø Alle forsøgsdata og et sæt dokumenter Ø Offentligt register med højere transparens Ø En mere strømlinet ansøgningsproces og overvågning i medlemsstaterne, koordineret vurdering af multinationale forsøg og en afgørelse pr. medlemsstat. Ø Bindende tidsfrister for godkendelse af forsøg og stiltiende accept. Ø Kompliceret nationalt og europæisk samarbejde, der kræver forandringer hos LMST og etiske komitéer. Ø Konkurrence mellem medlemsstater i en global arena. 5
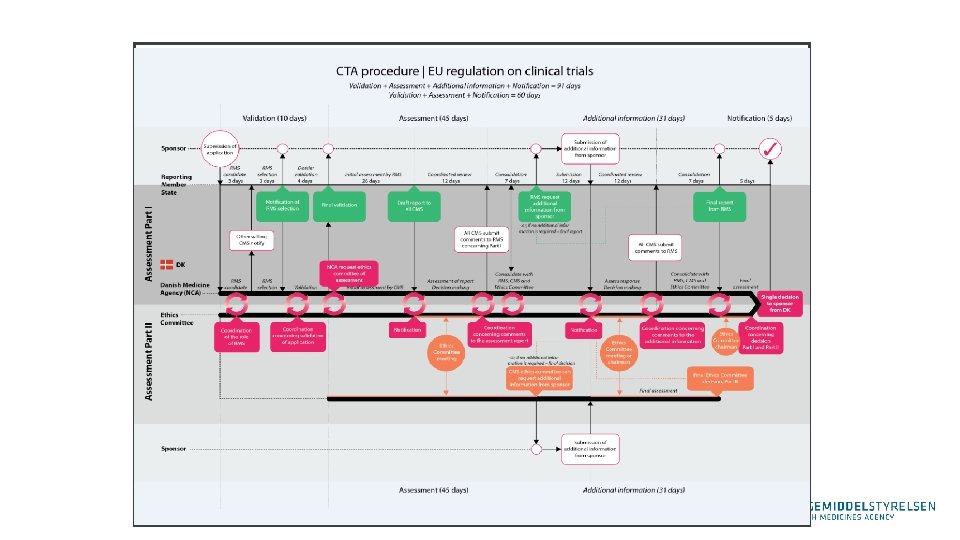
Timeline Submission Deadline misses Stiltiende accept Deadline misses ansøgning frafalder Validering Sponsor informeres om mangler Sponsor kan ikke svare tilfredsstillende Ansøgning trækkes Assesment Sponsor informeres validering OK Sponsor leverer manglende informeres Deadline misses Stiltiende accept Deadline misses ansøgning frafalder Sponsor informeres om mangler Sponsor kan ikke svare tilfredsstillende Ansøgning trækkes Sponsor informeres om afgørelse Sponsor leverer manglende informeres
Forskellen på direktiv 2001/20/EU og forordning ‒ ”Low interventional trials” – IMP er markedsført og bruges jf. markedsføringtilladelsen el. brugen er evidens-baseret i Member State – Diagnostiske el. monitorerings procedurer udgør en minimal ekstra risici ‒ NIMP = auxiliary (hjælpe-) medicinal products – disse falder nu indenfor anvendelsesområde! – AMP skal være markedsført. Sm. PC medsendes hvis MT er fra andet EU land og kræver begrundelse og documentation hvis AMP ikke har MT ‒ Øget transparens omkring kliniske forsøg - alt i EU databasen skal offentliggøres ‒ Forenklet sikkerhedsrapportering med samarbejde mellem Member States – Non-serious AE og AR er ikke dækket af forordning. ‒ Krav til etikettering, ikke længere bare en guideline
Part I (EU) Cover letter EU application form Protocol Investigator’s Brochure IMPD GMP compliance AMP Dossier Scientific advice/PIP Labelling One CT dossier Part II (MS) Recruitment arrangements Subject info/informed consent Suitability of investigator Suitability of facilities Proof of insurance Financial /other arrangements Proof of payment of fee EU Law on Data protection EU Portal
Hvad skal ansøgningen indeholde ‒ Forordningens Bilag 1 – Ansøgningsdossier vedr. ansøgning ‒ EU-portal – Ansøger skal registreres – Forsøget skal registreres med informationer a la Eudra. CT – Dokumenter skal uploades 9 27. OKTOBER 2020
Europæisk procedure for godkendelse ‒ Kun medlemslande hvor forsøget ønskes gennemført involveres. ‒ Referenceland udpeges af sponsor. ‒ Referenceland udarbejder vurderingsrapport. ‒ Koordination mellem medlemslande ved vurdering. ‒ Klart definerede tidsrammer (korte) og stiltiende accept i mangel af information. ‒ Mulighed for ”opt out” hvis betydelige forskelle i klinisk praksis eller forsøg i uoverenstemmelse med national lov (videnskab og socioøkonomi). ‒ Single opinion pr. land
EU-portal For at understøtte forordningen skal der laves en EU-portal og EU-database Forordningen implementeres ikke før 6 måneder efter en audit af EU-portalen og EUdatabasen – nuværende oktober 2018 EU-portalen og –databasen udvikles af EMA i samarbejde med myndigheder og stakeholders 27 OKTOBER 2020
Programme governance MS, EMA & Commission Management Board MS representatives Stakeholders EU MSs, IT, Commission EMA Sponsors (commercial and not commercial organisations) Programme Managers IT Directors Executive Committee Project Teams EU TMB CT System Expert Group* Subgroups (delegates from CT System Expert Group) CROs Sponsor driven CT dossier submission process (including CT dossier creation) (LGP) Patient organisations Member state driven Validation, assessment and Authorization process Process Workflows (MARE) Health Care Professionals Clinical trial safety reporting (MARE) Inspection (PLM) Public access Provision to public, patient groups and others Manage user access Registration process and user management User roles and access rights EU controls and EC 12 * Continuing group first convened by the Commission in 2013 Taskforce engagement from DK: • Assessement reports part I og II • RMS selection process • Transperancy
System overview Interface with MSs CT systems 13
EMA’s tidsplan for udvikling af EU database og portal 14
Hvad gør vi Danmark? 15 27. OKTOBER 2020
Konklusion af mapping af processen Koordination mellem LMST og Etisk komite er essentielt Ø Møde i Videnskabsetiske lægemiddelkomiteer (VLK) hver 12 dag Ø Dedikerede og trænede medarbejdere som varetager koordinationen mellem LMST og VLK Ø Kommunikation på engelsk Ø IT platform til at udveksle information på national niveau for at understøtte en effektiv proces Ø Endnu ikke helt klarlagt hvordan EU-database og portalen præcist understøtte den nationale proces. 17
Forberedelse i DK ‒ Lægemiddelstyrelsen og Den nationale Videnskabsetiske Komité (DNVK) er i gang med for-analyse om evt. national IT-system ‒ Der skal være tæt samarbejde mellem LMST og VEK, da VEK også skal vurdere protokollen i del I ‒ Lov om kliniske forsøg med lægemidler er vedtaget – Udarbejdelse af bekendtgørelse som beskrevet i loven 18
Arbejdsgrupper under Sundhedsministeriet Stakeholders Coordination group Danish Ministry of Health, The national committee on Health Research Ethics Danish Medicines Agency IT support Danish Medicines Agency The national committee on Health Research Ethics Legislative issues and processes Danish Ministry of Health, Danish Medicines Agency, The national committee on Health Research Ethics Financing/fees Danish Ministry of Health Danish Medicines Agency The national committee on Health Research Ethics
Volumes (LMST data) Kapacitit: Ø 18 nye ansøgninger hver uge (stor variation) Ø Work in progress: 80 CTA’s og 40 amendments – i alt 120! Ca. 300 nye forsøg hvert år Ø Ca. 200 forsøg forventes at være multinationale (RMS or CMS)/Ca. 100 forsøg forventes at være nationale (RMS) Ø 180 forsøg fra commercial sponsors/120 forsøg fra non-commercial sponsors Højt antal af additional submissions Ø Ca. 700 amendments hvert år Ø Ca. 1500 -2000 additional submissions (safety reports, SUSAR, notifications) Volumes forventes at stige – part I og part II submissions for sig. 20
Lov om kliniske forsøg med lægemidler Lovforslag vedtaget 10. maj 2016 Hvad trådte i kraft 1. juli 2016: • direkte adgang • Komitéloven og Lægemiddelloven er ændret pr. 1. juli 2016 • Samtykket giver sponsor og sponsors repræsentant og investigator direkte adgang til at indhente oplysninger i patientjournaler mv. herunder elektroniske journaler…. . som led i bl. a monitorering. • Gælder ikke for udenlandske myndigheder – dvs. fuldmagt skal stadig bruges 27 OKTOBER 2020
Lov om kliniske forsøg med lægemidler • Nedsættelse af Videnskabsetiske lægemiddelkomiteer. • Beskriver samarbejde mellem Videnskabsetiske lægemiddelkomiteer (VLK) og Lægemiddelstyrelsen i bemærkningerne • Del I af vurderingsrapporten skal vurderes både af VLK og LMST • Del II af vurderingsrapporten skal kun vurderes af VLK 27 OKTOBER 2020
Kompetencer (Myndigheder i Danmark) Ø National Contact Point – Lægemiddelstyrelsen (LMST) Ø Faciliterer procedurerne i forordningen, kapitel II og III Ø Medlem af Clinical Trial Advisory Group (CTAG) Ø LMST håndterer opgaver/procedurer når Danmark er reporting member state (RMS) Ø LMST vurderer om et forsøg er omfattet af the scope of the regulation Ø Forpligtigelse for etisk komite og LMST at samarbejde ved assessment af applications og notifications 23
Assessment Report part I og II ‒ LMST vurderer part I (sundhedsfaglige aspekter) ‒ Etisk Komité vurdere part I (videnskabsetiske aspekter) og part II ‒ Substantial modification + DK som additional MS: – Både etisk Komité og LMST deltager i godkendelses processen – samme procedure som for nye forsøg (se ovenfor) ‒ Stadig to uafhængige myndigheder og beslutninger: – Single opinion pr. land – men: – Hvis VLK ikke enig i del I, kan DK ikke give tilladelse til forsøget – Hvis VLK ikke kan godkende del II, kan DK ikke give tilladelse til forsøget 24
Overgangsordning Når forordningen finder anvendelse (oktober 2018) kan sponsor vælge • At bruge EU-portalen m. v. • At bruge muligheden for at vente op til 1 år, hvor man så søger jf. direktiv 2001/20/EU • Forsøg, der har fået tilladelse efter direktiv 2001/20/EC, kan blive håndteret efter dette direktiv indtil 3 år efter at forordningen finder anvendelse • Herefter overgår det til forordningen 27 OKTOBER 2020
Hvad gælder nu for lægemiddelloven? Følgende er stadig gældende for kliniske forsøg: ü Lov om lægemidler §§ 88 -92 ü GCP-bekendtgørelse ü Klinisk forsøg bekendtgørelse Fra den nye Lov om kliniske forsøg med lægemidler Følgende trådte i kraft 1. juli 2016: I § 89 i Lægemiddelloven indsættes nyt stk. 2 vedr. direkte adgang – se tidligere slide Resten af Lov om kliniske forsøg med lægemidler træder først i kraft når ministeren fastsætter tidspunkt (formentlig oktober 2018) 27 OKTOBER 2020
Forberedelse til forordningen For at forberede sig på forordningen kan man: • Bruge VHP – samlet vurdering af protokol, IB og IMPD (part I) • Forberede sig på at anmelde forsøg via DKMAnet – en samlet ansøgning for LMST og VEK – part I og part II i EU-portalen 27 OKTOBER 2020
Tak for opmærksomheden Tag evt. kontakt: Sektion for kliniske forsøg Tlf. nr. 4488 9123 E-mail: kf@dkma. dk 27 OKTOBER 2020