Postprocessing long pairwise alignments 93428 Zheng Zhang et














 Search Time: n n If k such")



































- Slides: 54
Post-processing long pairwise alignments 陳啟煌 93/4/28 Zheng Zhang et al. , Bioinformatics Vol. 15 no. 12 1999
Outline n n Motivation Theoretical basis of the proposed algorithms How to build up Useful Tree An application
Motivation n Avoid local alignment problems n n Smith-Waterman lead to inclusion of an arbitrarily poor internal segment. Others approaches may generate an alignment score less than some internal segment
Smith-Waterman approach C 0 G G A T C A 0 0 0 0 C 0 T 0 8 5 2 0 0 8 5 5 3 0 0 8 5 3 13 0 2 0 0 0 8 5 2 11 0 0 8 5 3 13 10 0 0 8 5 2 11 8 0 8 5 2 5 3 13 10 7 0 5 3 0 2 13 10 8 18 0 T A A C T T 2 The best score
Inclusion of a poor segment n Inclusion of an arbitrarily poor region in an alignment n Smith-Waterman approach potential flaws.
X-Alignments n n n An X-Drop within an alignment, where X>0 is fixed in advance. A region of consecutive columns scoring less than <-X Alignments contain no X-Drop, we call X-alignments
BLAST In Blast Step 3: Extend hits. hit Terminate if the score of the extension fades away. (That is, when we reach a segment pair whose score falls a certain distance below the best score found for shorter extensions. )
2 X-Drop
Non-normal alignment The HSP has been extended to the right side in such a way that the entire alignment score less than the section from a to b
The Proposed Approach n Provide techniques for decomposing a long alignment into sub-alignments that avoid the both problems. n n Show to scan an alignment to collect information from which a decomposition corresponding any X can be found almost instantaneously. Provide a method for detecting variations in the rate of genome evolution
Useful Tree
X-full alignment n n n An alignment are normal if each of its prefixes or suffixes has non-negative score. An alignment is not contained in any longer normal alignment is called full X-alignment + maximal X-normal is called X-full
X-full alignment n n 0 -full alignment is maximal runs of columns of A with non-negative scores. For every X, X-full alignments are pairwise disjoint. If X<Y X-full alignment contained in Yfull alignment. -full alignments are just full alignments
Useful Tree n n Encode X-full alignments for all X≥ 0 in tree data structure. Leaves: 0 -full alignments & maximal runs of negative score columns alternately Terminal Leaves: add two special leaves with score - Each internal node is a disjoint union of its three children. Keep alignment’s score and the minimum sub-alignment’s score
Time complexity n n Construct time: O(N) Search Time: n n If k such alignments, need inspect at most 3 k+1 nodes (2 k+1) leaves+((2 k+1 -1)/2) internal nodes =3 k+1 nodes
Decompose rules n n n Alignment A A 1, A 2, …. , A 2 n-1 # of sub-alignment is odd i : score of Ai Negative & Non-negative score alternately 0= 2 n= -∞
Theoretical basis
Theoretical basis
Theoretical basis n n Lemma 1: X is consistent Lemma 2: A normal drop is consistent with X
Lemma 3
Useful tree definition n Each node of T is a segment consistent with X. Each leaf of T is of the form [i, i+1) Each internal node [a, d) has exactly three children. [a, b), [b, c) and [c, d) and the signs of their scores alternate.
Lemma 4
Possible negative merge n LEMMA 5. Assume that three consecutive roots in our sequence, [a, b), [b, c), and [c, d), satisfy n n n 0 ≤ (b, c)< min(- (a, b), - (c, d)) Then merging these trees into a single tree with root [a, d) creates a useful tree and the resulting sequence still satisfies P 1 and P 2. If a, b, c and d satisfy this lemma, [a, d) is a possible negative merger.
Possible positive merge n LEMMA 6. Assume that five consecutive roots in our sequence, [a, b), [b, c), [c, d), [d, e) and [e, f) satisfy n n 0 > (c, d) ≥ max( (a, b), (e, f)) neither [a, d) nor [c, f) is a possible negative Then merging these trees into a single tree with roots[b, c), [c, d), [d, e)into a single root[b, e) creates a useful tree and the resulting sequence still satisfies P 1 and P 2. If a, b, c, d, e and f satisfy this lemma, [a, d) is a possible positive merger.
Lemma 7
Theoretical basis n n n Normal rise and normal drop Useful Tree contains every segments Possible negative merger Possible positive merger Always exists possible negative merger or possible positive merger
Decompose rules n n n Alignment A A 1, A 2, …. , A 2 n-1 # of sub-alignment is odd i : score of Ai Negative(odd i) & Non-negative(even i) score alternately 0= 2 n= -∞
Useful Tree build up procedure 1. Push the first leaf on the stack 2. While the stack size exceeds 1 or there is an unvisited leaf do 3. if the top three stack items indicate a negative merger then 4. 5. 6. 7. 8. pop three items, merge them and push the result onto the stack else if the top five segments indicate a positive merge then pop an item{e, f} perform line 4. and push {e, f} back else push the next two leaves onto the stack
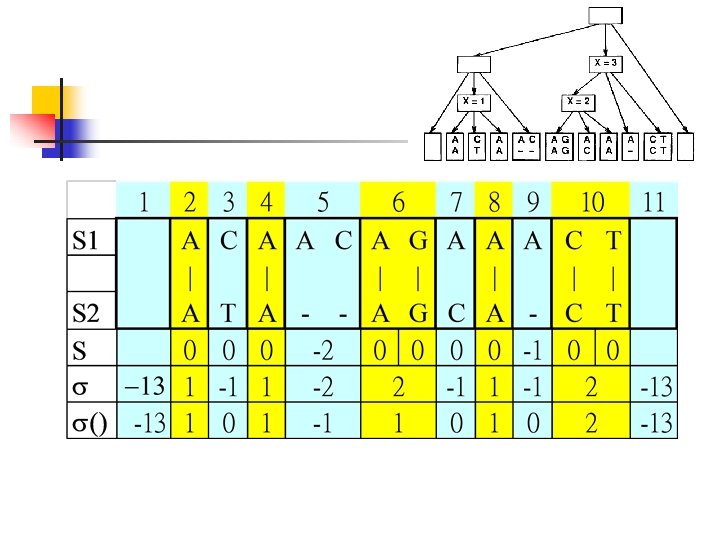
Construct Useful Tree n n n ACAACAGAAACT | | || ||| ATA--AG-CACT Gop: 0 Gep: 1 Match/mismatch: 1/-1
n n n Push 1 Push 2, 3 Push 4, 5, n n Push 6, 7 Push 8, 9 n n Merge 2, 3, 4 as a Merge 1, a, 5 as b Merge 6, 7, 8 as c Push 10, 11 n n Merge 9, 10, 11 as d Merge b, c, d as e
n Source code of this paper n http: //globin. cse. psu. edu/dist/decom/
Alignment file n n n n #: lav d{ "simu elegans briggsae M = 10, I = -10, V = -10, O = 60, E = 2" } s{ "s 1" 1 12 "s 2" 1 9 } h{ ">SUPERLINK_RWXL 2782216 -2889703" ">dna -c briggsae. dna " } a{ n s 562 n b 1 1 n e 3 3 n l 1 1 3 3 99 n l 6 4 9 7 99 n l 11 8 12 9 99 n} n
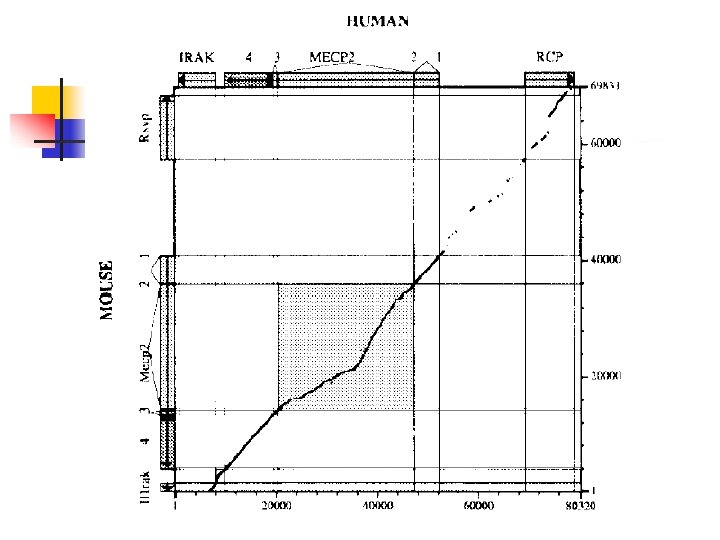
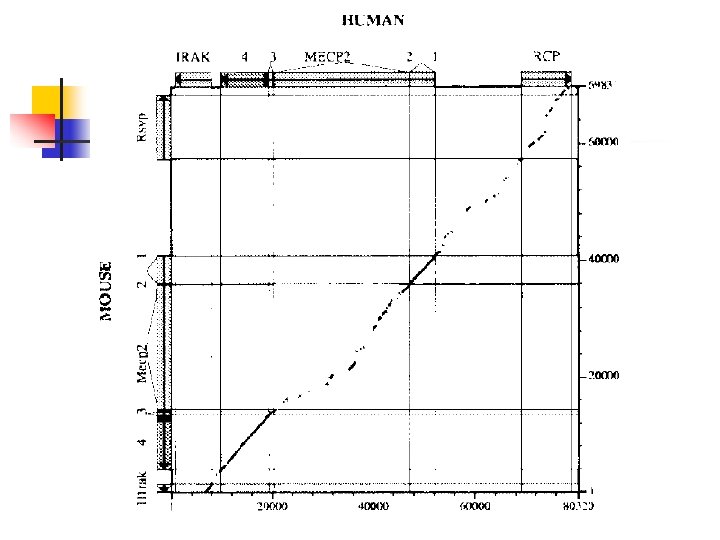
An Application n Different regions of a mammalian genome evolve at different rates. Provide a method for detecting variations in the rate of genome evolution To compare the rates of evolution in different genomic regions from humans and mice. n Align each pair of homologous regions and determined
Pitfalls n n Tally statistics only at sequence not in exons Regions adjacent to an exon maybe be aligned n n Remove the exons before producing the alignment The alignment program is unable to differentiate the biologically meaning alignment
Proposed approach n n First align the sequences using the exons as guideposts Then re-score the alignment where positions within exons are masked, so that they cannot be aligned to another nucleotide.
References n n n Zheng Zhang et al. , “Post-processing long pairwise alignments”, Bioinformatics, Vol. 15 no. 12 1999 http: //globin. cse. psu. edu/dist/decom/ Kun-Mao Chao , Algorithms for Biological Sequence Analysis Lecture Notes, National Taiwan University, Spring 2004
Q&A n Thank you!
Possible mistakes, but maybe not n n n P. 1015 left col. , last 2 row ∑ k=1 ∑ k=i P. 1015 Right col. [i, i) should be [i, j) P. 1016 proof of lemma 4 4 [i, i) should be [i, j) P. 1017 proof of lemma 5 (b, c) (e, c) should be (b, c)- (e, c) P. 1017 lemma 7 (ai-3, ai-2) (ai-4, ai-1)
n Lemma 1: X is consistent Proof 1
Proof of lemma 2
Lemma 3
Proof of lemma 3
Lemma 4
Proof of lemma 4
Possible negative merge n LEMMA 5. Assume that three consecutive roots in our sequence, [a, b), [b, c), and [c, d), satisfy n n n 0 ≤ (b, c)< min(- (a, b), - (c, d)) Then merging these trees into a single tree with root [a, d) creates a useful tree and the resulting sequence still satisfies P 1 and P 2. If a, b, c and d satisfy this lemma, [a, d) is a possible negative merger.
Proof of lemma 5
Possible positive merge n LEMMA 6. Assume that five consecutive roots in our sequence, [a, b), [b, c), [c, d), [d, e) and [e, f) satisfy n n 0 > (c, d) ≥ max( (a, b), (e, f)) neither [a, d) nor [c, f) is a possible negative Then merging these trees into a single tree with roots[b, c), [c, d), [d, e)into a single root[b, e) creates a useful tree and the resulting sequence still satisfies P 1 and P 2. If a, b, c, d, e and f satisfy this lemma, [a, d) is a possible positive merger.
Proof of lemma 6
Lemma 7
Proof of lemma 7