Mechanizmy reakcji organicznych 1 Jak rozpocz pisanie mechanizmu














 • Ustal strukturę każdego reagenta.")


 jest dany atom wodoru")


 • reakcja faworyzowana lub utrudniona Reakcja jest uprzywilejowana gdy")




 Reakcje silnie egzotermiczne")


![Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • [4+2] cykloaddycja W niższej temperaturze powstaje](https://slidetodoc.com/presentation_image/28d442a82c1daa293bd726b3ab7a94d9/image-38.jpg "Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • [4+2] cykloaddycja W niższej temperaturze powstaje")






















- Slides: 66
Mechanizmy reakcji organicznych
1. Jak rozpocząć pisanie mechanizmu reakcji 2. Reakcje polarne zachodzące w warunkach zasadowych 3. Reakcje polarne w warunkach kwaśnych 4. Reakcje pericykliczne 5. Reakcje wolnorodnikowe 6. Reakcje promowane lub katalizowane metalami 7. Metody badania mechanizmów reakcji
1. Jak rozpocząć pisanie mechanizmu reakcji • Obrazowanie mechanizmów reakcji za pomocą strzałek • Kwasowość i zasadowość Brønsteda • Kinetyka i termodynamika (profile energii) • Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • Klasyfikacja przemian w chemii organicznej • Klasyfikacja mechanizmów reakcji chemicznych
Chemia organiczne jest dziedziną nauki która bada jak substancje organiczne są przekształcane od substratów w produkty
Mechanizmy reakcji organicznych są opisami dróg przekształcania substratów w produkty. Cząsteczki z takimi samymi grupami funkcyjnymi reagują wedle podobnych mechanizmów. Na podstawie mechanizmów znanych reakcji można przewidzieć zachowanie się podobnych cząsteczek w analogicznych warunkach. Mechanizmy reakcji są więc tym co łączy podobne reakcje. Znając je, można wyjaśnić sposób powstawania produktów znanych reakcji, a także przewidzieć reaktywność cząsteczek organicznych.
Science 31 Vol. 317 no. 5842 pp. 1189 -1192
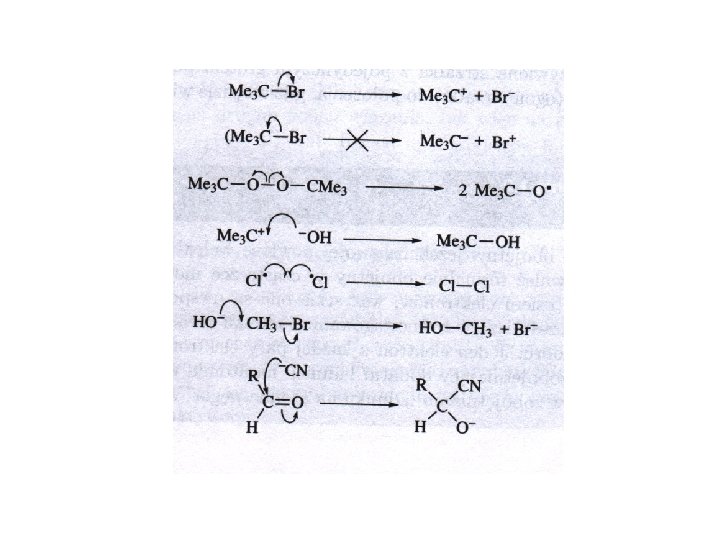
Obrazowanie mechanizmów reakcji za pomocą strzałek Wygięta strzałka przedstawia ruch pary elektronowej z orbitalu zapełnionego na pusty Gdy nukleofil atakuje niewiążący orbital jedna strzałka obrazuje powstawanie nowego wiązania, a druga pęknięcie starego
Zachowanie ładunku na każdym etapie reakcji Elektrony mogą być dostarczane również z wiązania π. Strzałka zaczyna się w środku wiązania π.
Zachowanie ładunku na każdym etapie reakcji
Rozpad cząsteczek Spontaniczny rozpad cząsteczek - przyczyna – słabe spolaryzowane wiązanie σ Grupy funkcyjne, które „oddalają się” z elektronami z wiązania σ (Br, N 2+, OH 2+) są zwane grupami opuszczającymi, a tego typu rozpad określamy jako rozczepienie heterolityczne Homolityczne rozczepienie – tworzenie rodników
Przemieszczanie elektronów wewnątrz cząsteczki
Podsumowanie. Co oznaczają wygięte strzałki: • Wygięta strzałka pokazuje ruch pary elektronowej • Początek strzałki wskazuje źródło pary elektronowej, którym może być zapełniony orbital HOMO lub wolna para elektronowa wiązania π lub σ. • Grot strzałki pokazuje miejsce przemieszczenia pary elektronów, którym może być elektroujemny atom, który w wyniku tego przemieszczenie zyskuje ładunek ujemny (grupa opuszczająca), pusty orbital, gdy tworzone jest nowe wiązanie lub niewiążący orbital (σ* lub π*), gdy wiązanie jest przerywane. • Zachowany jest sumaryczny ładunek.
Wskazówki do samodzielnego pisanie mechanizmów reakcji (Clayden str. 131) • Ustal strukturę każdego reagenta. • Ustal, który atom jest nukleofilowy, a który elektrofilowy. • Zaznacz wolną parę na nukleofilowym atomie. • Wygięte strzałki zawsze rysuj w tym samym kierunku. • Jeśli tworzysz nowe wiązanie z H, C, N lub O musisz zerwać już istniejące wiązanie. • Zaznacz wyraźnie ładunki w reagentach i produktach pośrednich. • Upewnij się, że ładunek całkowity w Twoim mechanizmie jest zachowany.
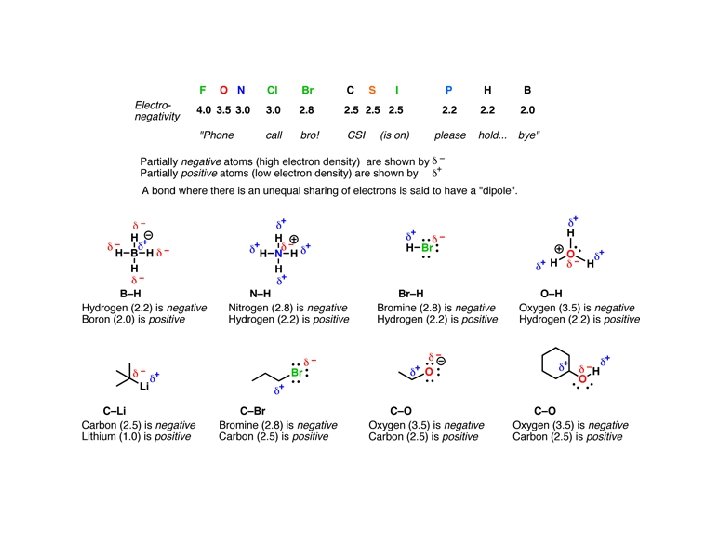
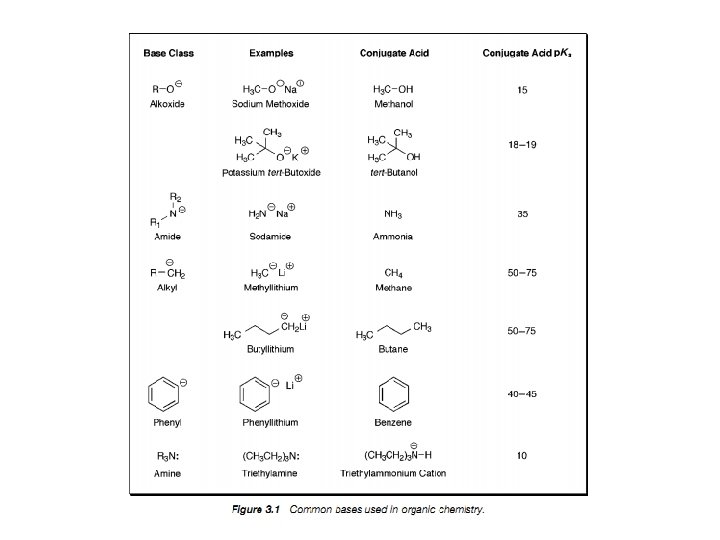
Kwasowość i zasadowość Brønsteda • Kwasem jest substancja wykazująca skłonność do utraty protonu • Zasadą jest substancja wykazująca skłonność do przyłączenia protonu • Zasady mogą być anionowe lub neutralne, a kwasy neutralne bądź kationowe • Reakcje kwasowo zasadowe są reakcjami równowagowymi • Kwas i zasadę mamy po obu stronach reakcji równowagowej • Reakcja transferu protonu jest reakcją bardzo szybką szczególnie gdy proton jest transferowany z jednego heteroatomu na drugi
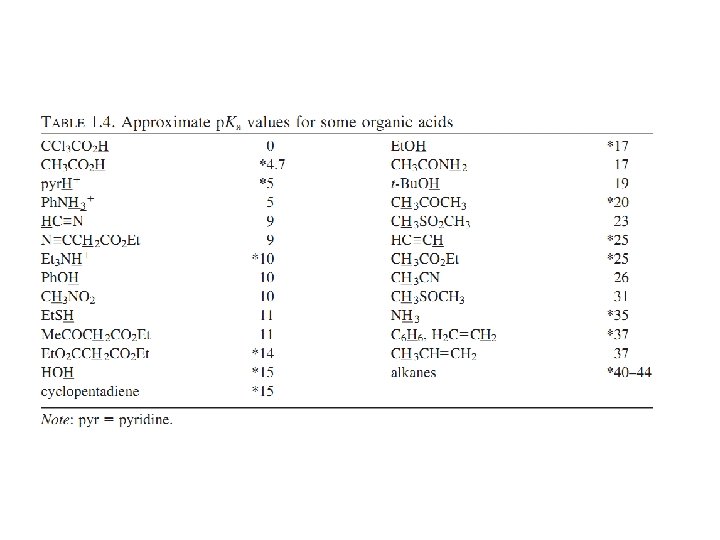
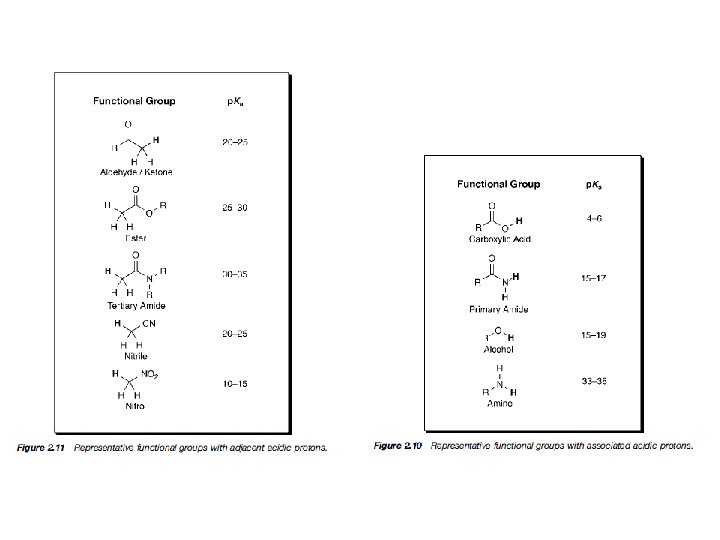
Wartość p. Ka określa, jak bardzo kwasowy (lub jak mało) jest dany atom wodoru w rozpatrywanym związku. Jest to bardzo użyteczne, ponieważ w sytuacji, gdy pierwszym etapem reakcji jest protonowanie lub deprotonowanie jednego z substratów, trzeba wiedzieć, w którym miejscu związek ten może być protonowany bądź deprotonowany i jak mocny kwas lub zasada będzie tu potrzebna.
Do zapamiętania: • Kwasowość wzrasta wraz ze wzrostem elektroujemności (porównaj: H 3 CH, H 2 NH, HOH) • Kwasowość wzrasta idąc w dół układu okresowego (porównaj: Et. OH z Et. SH). Jest to prawdopodobnie spowodowane słabym nakładaniem się małych orbitali wodoru z rosnącymi walencyjnymi orbitalami atomów, z którymi jest połączony. • Kwasowość związku HA wzrasta kiedy indukcyjnie elektronoakceptorowe grupy są połączone z A , a maleje gdy grupy elektronodonorowe są połączone z A (porównaj CCl 3 COOH z CH 3 COOH, i HOH z Et. OH). • Dla neutralnych kwasów, kwasowość maleje ze wzrostem zawady przestrzennej (porównaj Et. OH, t-Bu. OH). • Kwas (HA) jest zdecydowanie bardziej kwaśny kiedy wolna para sprzężonej zasady jest stabilizowana przez rezonans (porównaj: Ph. OH z Et. OH i CH 3 CH=CH 2 z alkanami). HA jest wyjątkowo kwaśny gdy wolna para jest delokalizowana z jedną lub dwoma grupami karbonylowymi.
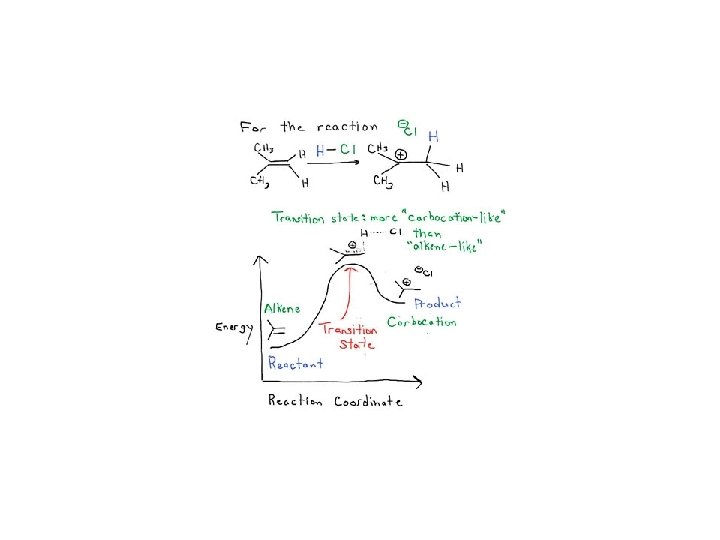
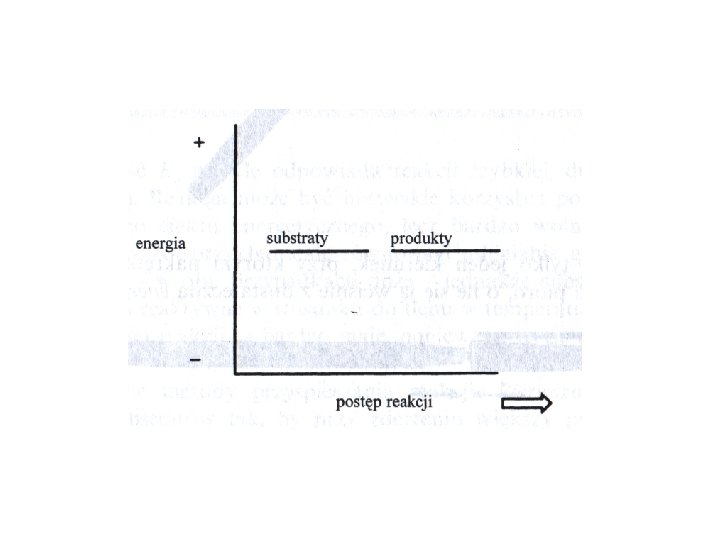
Kinetyka i termodynamika (profile energii) • reakcja faworyzowana lub utrudniona Reakcja jest uprzywilejowana gdy energia swobodna jest mniejsza od zera. Energia swobodna jest zależna od entalpii (ΔH) i entropii (ΔS), ΔGo= ΔHo -TΔSo. W praktyce to entalpie nie energie swobodną wykorzystuje się do określenia czy reakcja jest preferowana czy nie. Reakcja z ΔHo < 0 jest egzotermiczna, ΔHo > 0 endotermiczna.
Kinetyka i termodynamika • reakcja szybka czy wolna Substraty po przekroczeniu bariery energetycznej przechodzą w produkty. Energia potrzebna by substraty osiągnęły szczyt bariery energetycznej to energia aktywacji, a szczyt bariery, w którym reagenty mogą równie łatwo wrócić do substratów lub przejść do produktów nazywamy stanem przejściowym. Szybkość reakcji jest zależna od wielkości bariery aktywacji.
Kinetyka i termodynamika • stan przejściowy
Kinetyka i termodynamika • stan przejściowy
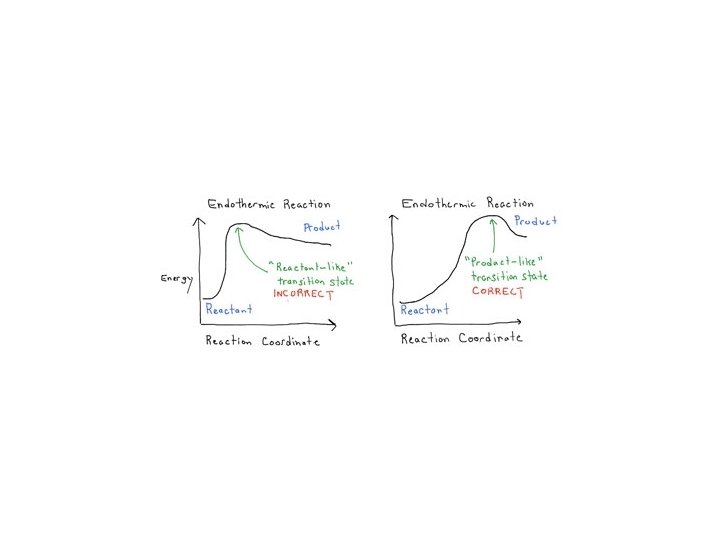
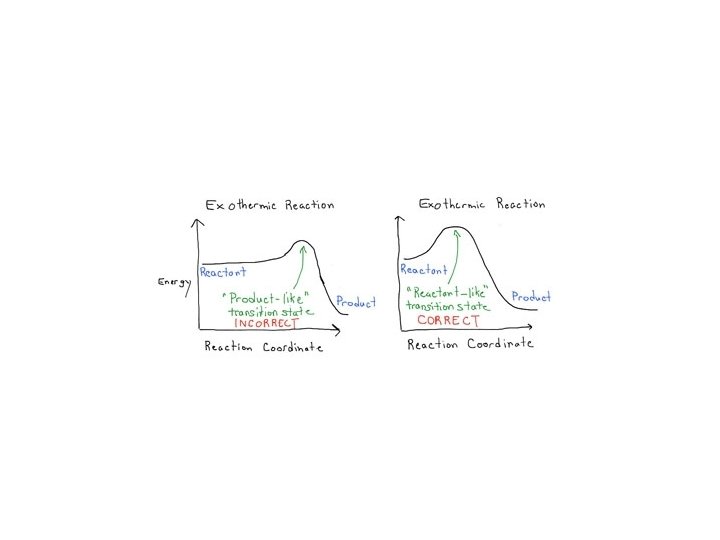
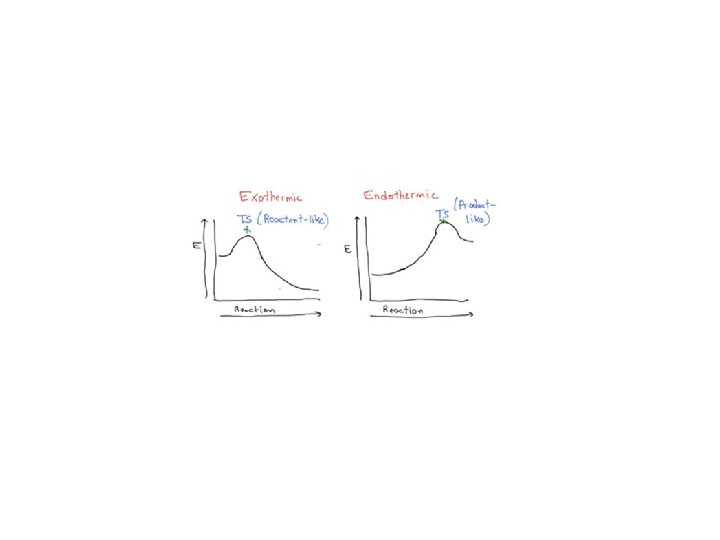
Kinetyka i termodynamika • postulat Hammonda Jeżeli dwa stany, na przykład stan przejściowy i niestabilny związek pośredni, następują kolejno podczas procesu reakcji i mają prawie taką samą zawartość energii, ich wzajemna konwersja będzie obejmować tylko niewielką reorganizację struktur molekularnych.
Kinetyka i termodynamika • postulat Hammonda (wczesny i późny stan przejściowy) Reakcje silnie egzotermiczne charakteryzują się wczesnym stanem przejściowym i wykazują małą selektywność. Reakcje silnie endotermiczne charakteryzują się późnym stanem przejściowym i wykazują wysoką selektywnością.
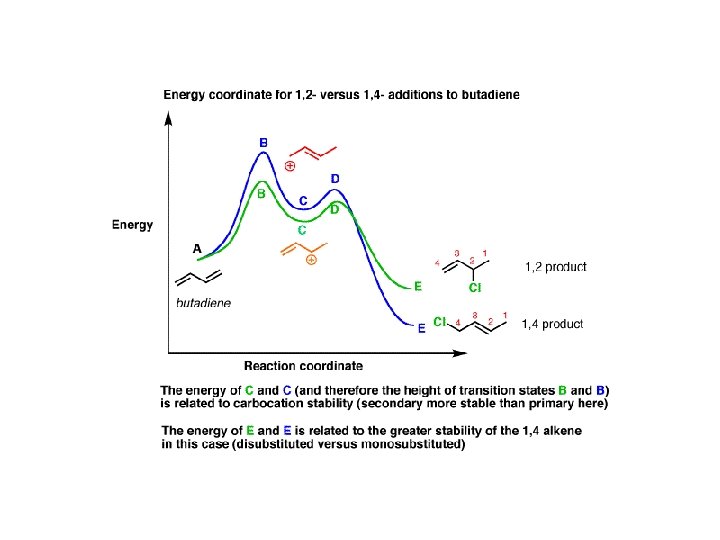
Na potencjalny wynik reakcji zwykle wpływają dwa czynniki: 1. względna stabilność produktów ( tj. współczynniki termodynamiczne) 2. szybkość tworzenia produktu ( tj. czynniki kinetyczne) Reakcja 1 ( zielona ) wytwarza produkt 1 ( P 1 ) za pośrednictwem stanu przejściowego 1 ( TS 1 ). Będzie to szybsza reakcja, ponieważ ma mniejszy (bardziej stabilny) stan przejściowy energii, a zatem niższą barierę aktywacyjną. Dlatego produkt 1, P 1 jest produktem kinetycznym (produkt, który tworzy się najszybciej). Reakcja 2 ( niebieska ) wytwarza produkt 2 ( P 2 ) przez stan przejściowy 2 ( TS 2 ). P 2 jest bardziej stabilnym produktem, ponieważ P 2 ma niższą energię niż P 1. Dlatego P 2 jest produktem termodynamicznym (bardziej stabilnym produktem).
Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • pierwsze doniesienie o kontroli kinetycznej i termodynamicznej
Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • [4+2] cykloaddycja W niższej temperaturze powstaje produkt endo 2, w wyższej produkt termodynamicznie trwalszy egzo 1. Produkt egzo posiada niższe zawady przestrzenne, a w stanie przejściowym prowadzącym do produktu 2 występuje skuteczniejsze nakładanie orbitali tworzących nowe wiązania.
Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • deprotonowanie niesymetrycznych ketonów
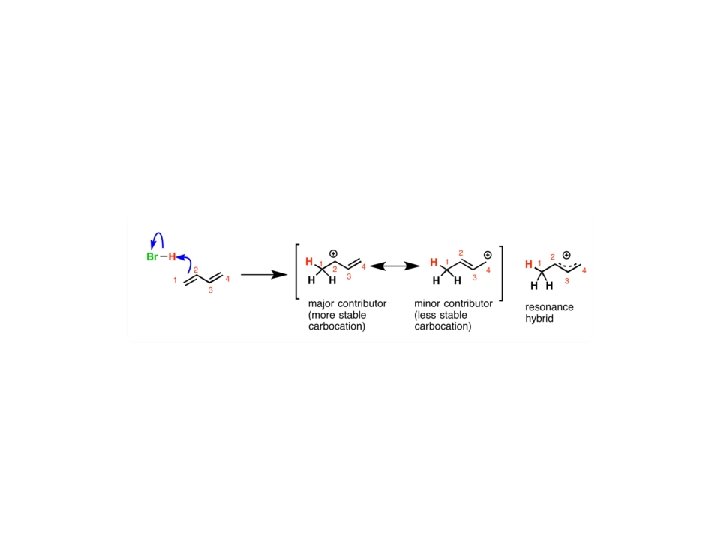
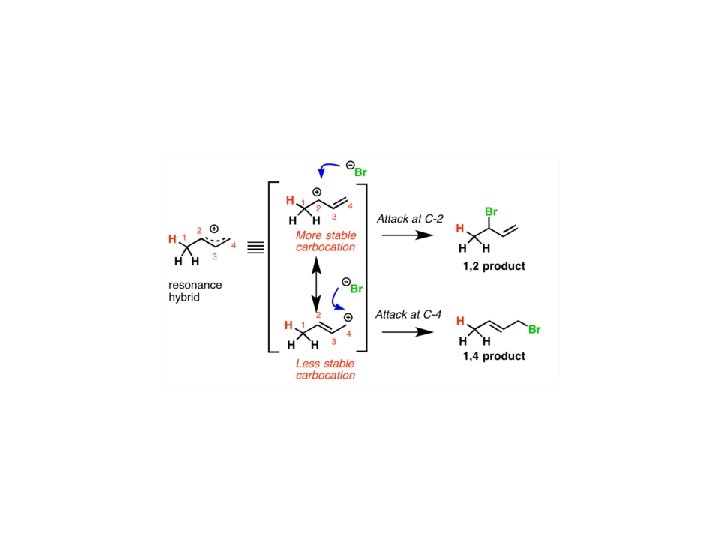
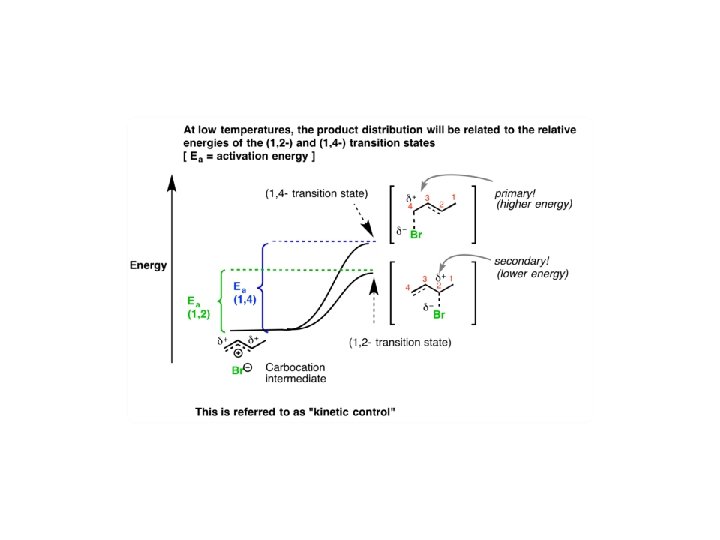
Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • addycja elektrofilowa
Klasyfikacja przemian w chemii organicznej • Reakcje addycji • Reakcje eliminacji • Reakcje substytucji • Reakcje przegrupowania
Klasyfikacja reakcji chemicznych • W reakcjach addycji dwa substraty łączą się dając jeden produkt. Zwykle wiązanie π, które jest jednym z substratów zostaje zastąpione dwoma wiązaniami σ
Klasyfikacja reakcji chemicznych • W reakcjach eliminacji jeden substrat ulega podziałowi na dwa produkty. Zwykle dwa wiązania σ w materiale wyjściowym zostają zastąpione przez nowe wiązanie π
Klasyfikacja reakcji chemicznych • W reakcjach substytucji atom lub grupa związana wiązaniem σ z resztą cząsteczki zostaje zastąpiona przez inny σ-związany atom lub grupę.
Klasyfikacja reakcji chemicznych • W reakcjach przegrupowania pojedynczy materiał wyjściowy daje jeden produkt o innej strukturze.
Klasyfikacja mechanizmów reakcji chemicznych • Reakcje polarne – procesy te polegają na ruchu par elektronowych z obszarów o wysokiej gęstości elektronowej (nukleofile) do obszarów o niskiej gęstości elektronowej (elektrofile), lub z pełnego orbitalu na orbital pusty. • Reakcje wolnorodnikowe polegają na ruchu pojedynczych elektronów. Nowe wiązanie powstaje wskutek połączenia dwóch połowicznie zapełnionych (half-filled) orbitali. • Reakcje pericykliczne charakteryzują się kołowym ruchem elektronów. • Reakcje promowane lub katalizowane metalami
Mechanizmy Polarne – reakcje nukleofili z elektrofilami biegną zwykle w kwaśnych lub zasadowych warunkach. • Nukleofile to związki posiadające wolną aktywną parę elektronową dostępną by wytworzyć nowe wiązanie. Nukleofil może być obojętny elektrycznie lub naładowany ujemnie. Istnieją trzy typy nukleofili: nukleofile posiadające wolna parę elektronową, σ-nukleofile i πnukleofile. • Nukleofile posiadające wolna parę elektronową. Wolna para jest wykorzystywane do tworzenia nowego wiązania z elektrofilem. Alkohole (ROH), aniony oksoniowe, aminy, aniony chlorkowe, tiole, fosfiny.
• σ-nukleofile – posiadają wiązanie między niemetalem a metalem. Elektrony tworzące wiązanie pierwiastkiem (niemetalem) a metalem zostają użyte do wytworzenia nowego wiązania między niemetalem, a elektrofilem. Formalny ładunek atomu nukleofilowego się nie zmienia, a metalu wzrasta o 1. Atomem nukleofilowym może być heteroatom (Na. NH 2, KOH), związki metaloorganiczne (zw. Grignarda, Gilmana, litoczy miedzioorganiczne) lub wodór jak w kompleksach wodorków Na. BH 4, Li. Al. H 4
• π-nukleofile – wykorzystują parę elektronową wiązania π do wytworzenie wiązania σ między jednym atomem z wiązania π, a atomem elektrofilowym. Formalny ładunek nukleofilowego atomu w wiązaniu π się nie zmienia lecz drugi atom zespołu π staje się ubogi w elektrony. Proste alkeny i areny są słabymi π nukleofilami podczas gdy wiązania π bezpośrednio połączone z heteroatomami (enaminy, enolany, etery winylowe) to aktywne π nukleofile.
Własności nukleofili • Nukleofilowość wzrasta posuwając się w dół układu okresowego, podczas gdy zasadowość maleje. Tak więc, I- jest świetnym nukleofilem podczas gdy Cl- słabym, Et 2 S to bardzo dobry nukleofil a Et 2 O bardzo słaby. • Nukleofilowość maleje dramatycznie ze wzrostem zawady przestrzennej wokół atomu nukleofilowego. • Delokalizacja ładunku zdecydowanie obniża zasadowość, a nieznacznie nukleofilowość. • Brak wiązania wodorowego w aprotycznych rozpuszczalnikach polarnych zwiększa zarówno nukleofilowość jak i zasadowość.
Elektrofile i grupy upuszczające. • Elektrofile są to związki posiadające niskoenergetyczny wolny orbital dostępny do wytworzenia nowego wiązania. Elektrofile mogą być obojętne lub posiadać ładunek dodatni. Mamy trzy typy elektrofili: elektrofile będące kwasami Lewisa, π elektrofile i σ elektrofile. • Elektrofile będące kwasami Lewisa posiadają atom E który nie ma oktetu elektronowego, a posiada niskoenergetyczny niewiążący orbital (zwykle p). Wolna para elektronowa nukleofila jest wykorzystywana przez E by wytworzyć nowe wiązanie i uzupełnić elektrony do oktetu.
Elektrofile i grupy upuszczające. • π elektrofile elektrofilowy atom E posiada oktet lecz jest połączony przez wiązanie π z atomem lub grupą, która może przyjąć parę elektronową
Elektrofile i grupy upuszczające. • σ elektrofile mają strukturę E-X. Elektrofilowy atom E posiada oktet ale jest połączony wiązaniem σ z atomem bądź grupą X zwaną grupą odchodzącą
Elektrofile i grupy upuszczające. • Grupy opuszczające można podzielić na dwie rodziny. Pierwsza z nich to grupy dobrze opuszczające, druga to grupy źle opuszczające. Skłonność danej grupy do przynależności do jednej z dwóch powyższych rodzin jest ściśle powiązana z p. Kb grupy.
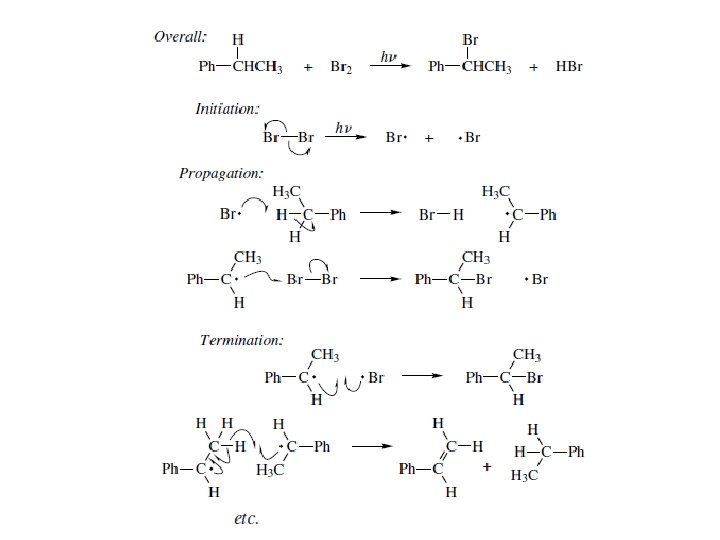
Reakcje wolnorodnikowe • Prawie wszystkie reakcje rodnikowe są reakcjami łańcuchowymi. Główne produkty reakcji powstają w etapie propagacji. Tylko niewielka ilość produktów ubocznych pochodzi z reakcji inicjacji i terminacji.
Reakcje pericykliczne • Reakcje perycykliczne powodują utworzenie lub zerwanie co najmniej jednego wiązania podwójnego. Często substraty lub produkty zawierają dwa sprzężone wiązania π. • Reakcje pericykliczne są stereospecyficzne. Czyli, że z trans wiązania podwójnego powstaje jeden diestereoizomer, a z izomeru cis drugi
Podsumowanie Jak zacząć pisać mechanizm reakcji? 1. Zaznacz ciężkie atomy w substratach i produktach 2. Zanotuj które wiązania σ uległy zerwaniu na drodze z substratów do produktów 3. Zaklasyfikuj sumaryczny przebieg reakcji (addycja, substytucja, eliminacja, przegrupowanie). Wiele procesów może składać się z dwóch lub więcej reakcji cząstkowych 4. Patrząc na warunki reakcji zakwalifikuj mechanizm (polarny w warunkach kwaśnych lub zasadowych, wolnorodnikowy czy promowany metalem) 5. Jeśli mechanizm jest polarny ustal nukleofilowość, elektrofilowość i kwasowość atomów gdzie wiązanie σ będzie tworzone lub zrywane.
Kontrola kinetyczna i termodynamiczna w reakcjach organicznych • Gdy w niskiej temperaturze powstaje jeden produkt, a w podwyższonej inny

 Elektrofile
Elektrofile Termodynamika
Termodynamika Anion wodorkowy + kation oksoniowy
Anion wodorkowy + kation oksoniowy Ustal wzor rzeczywisty alkoholu o masie czasteczkowej 62
Ustal wzor rzeczywisty alkoholu o masie czasteczkowej 62 Zaproponuj metode otrzymywania bromoetanu
Zaproponuj metode otrzymywania bromoetanu Psychologiczne mechanizmy uzależnienia
Psychologiczne mechanizmy uzależnienia Mechanizmy obronne wg vaillanta
Mechanizmy obronne wg vaillanta Mechanizmy ipc
Mechanizmy ipc Adresa na listoch
Adresa na listoch Skupinové číslovky
Skupinové číslovky Układanie zdań rozsypanki wyrazowe do wydruku
Układanie zdań rozsypanki wyrazowe do wydruku Písanie čísloviek slovom
Písanie čísloviek slovom Ako sa píšu číslovky slovom
Ako sa píšu číslovky slovom Zasady pisania rozprawki
Zasady pisania rozprawki Napisać zaproszenie
Napisać zaproszenie Písanie nôt
Písanie nôt Kuriuo paprastuoju mechanizmu galima laimėti darbo
Kuriuo paprastuoju mechanizmu galima laimėti darbo Skridiniai buityje
Skridiniai buityje Iso 4301-1
Iso 4301-1 Fungovanie trhového mechanizmu
Fungovanie trhového mechanizmu Nuozulniosios plokstumos ilgis
Nuozulniosios plokstumos ilgis Oblicz zmiane entalpii całkowitego spalania 1 mola propenu
Oblicz zmiane entalpii całkowitego spalania 1 mola propenu Alkohole monohydroksylowe ulegają reakcji
Alkohole monohydroksylowe ulegają reakcji Wydajność reakcji
Wydajność reakcji Entalpia reakcji wzór
Entalpia reakcji wzór Równowaga chemiczna reakcji 2so3
Równowaga chemiczna reakcji 2so3 Ustal wzór kwasu jednokarboksylowego jeżeli w reakcji 3 g
Ustal wzór kwasu jednokarboksylowego jeżeli w reakcji 3 g Mechanizm reakcji estryfikacji
Mechanizm reakcji estryfikacji Niemetal o symbolu s
Niemetal o symbolu s Entalpia reakcji
Entalpia reakcji Acuchu
Acuchu Zadania na szybkość reakcji
Zadania na szybkość reakcji Jak velkou silou se navzájem přitahují země a slunce
Jak velkou silou se navzájem přitahují země a slunce Jaka piękna jest warszawa
Jaka piękna jest warszawa Převod zlomků na desetinné číslo
Převod zlomků na desetinné číslo Na misiowe urodziny
Na misiowe urodziny Jak obliczyć moubr 2020
Jak obliczyć moubr 2020 Przykładowe opowiadanie
Przykładowe opowiadanie Jak sestrojit osu trojúhelníku
Jak sestrojit osu trojúhelníku Uzupełnij ustnie zdania brakującymi czasownikami
Uzupełnij ustnie zdania brakującymi czasownikami Wygląd orłów z hobbita
Wygląd orłów z hobbita Podmt
Podmt Biała droga rozkoszy
Biała droga rozkoszy Jak funguje trh
Jak funguje trh Vzor doporučeného dopisu
Vzor doporučeného dopisu Jak napsat adresu na doporučený dopis
Jak napsat adresu na doporučený dopis Procenty a punkty procentowe
Procenty a punkty procentowe Jak vyrobit model kuželu
Jak vyrobit model kuželu Jak zrobić dopalacze z tytoniu
Jak zrobić dopalacze z tytoniu Sinistrogram
Sinistrogram Szanuj bliźniego swego jak siebie samego
Szanuj bliźniego swego jak siebie samego Zapojení zásuvky 230v
Zapojení zásuvky 230v Jak odczytać cyfry rzymskie
Jak odczytać cyfry rzymskie Jak rozpoznać azbest
Jak rozpoznać azbest Vyjádři poměr v základním tvaru
Vyjádři poměr v základním tvaru Wilec ziemniaczany rozmnażanie
Wilec ziemniaczany rozmnażanie Przyspieszenie ziemskie w polsce
Przyspieszenie ziemskie w polsce Efa 14b
Efa 14b Jak byli traktowani izraelici w egipcie
Jak byli traktowani izraelici w egipcie Pit-37 wypełniony
Pit-37 wypełniony Jak sprzedawać produkty forever
Jak sprzedawać produkty forever Jak obliczyć moc silnika elektrycznego
Jak obliczyć moc silnika elektrycznego Vedení evidence hnojení
Vedení evidence hnojení Jak to zrobić
Jak to zrobić Jak zbudowany jest e papieros
Jak zbudowany jest e papieros Věta jednoduchá a souvětí
Věta jednoduchá a souvětí Rodzaje komputerów prezentacja
Rodzaje komputerów prezentacja


















