Bilanse Przykad 1 W kolumnie destylacyjnej rozdziela si

Bilanse

Przykład 1. W kolumnie destylacyjnej rozdziela się 1000 kg/h mieszaniny zawierającej 50% mas. benzenu, resztę stanowi toluen. Destylat zawiera 98, 5% benzenu, ciecz wyczerpana 3, 2% benzenu. Stosunek orosienia wynosi 0, 585. Sporządzić bilans pracy kolumny. Uwaga: dla zwiększenia efektywności destylacji frakcyjnej część destylatu jest zawracana do kolumny (zwykle na szczyt) w postaci tzw. orosienia. Przez stosunek orosienia rozumie się stosunek natężenia przepływu strumienia zawracanego do kolumny (orosienia) do natężenia przepływu destylatu.

W układzie kolumny destylacyjnej można wyróżnić dwa obszary bilansowe A i B. Do obszaru A strumienie dochodzą z zewnątrz (może być ich więcej, gdy są jeszcze inne strumienie zasilające) i wychodzą na zewnątrz. W obszarze B możemy wyróżnić podsystem, który grupuje strumienie wewnętrzne. Strumień odbierany ze szczytu kolumny rozdziela się na destylat (F 4) i orosienie (F 5). Schemat bilansowy rozdziału benzenu i toluenu w kolumnie destylacyjnej: 1 benzen, 2 – toluen

Jeżeli rozdzielacz orosienia potraktować jako samodzielną jednostkę procesową, to dla kolumny można napisać dwa równania bilansowe dla obydwu składników mieszaniny i dwa dla rozdzielacza sporządza się dwa bilanse: dla obszaru A i dla obszaru B. Dla kolumny: F 1 x 11 = F 4 x 41 + F 3 x 31 F 1 x 12 = F 4 x 42 + F 3 x 32 (1) (2) Dla rozdzielacza: F 2 x 21 = F 4 x 41 + F 5 x 51 F 2 x 22 = F 4 x 42 + F 5 x 52 Ponieważ strumienie F 2, F 4 i F 5 mają taki sam skład to te dwa równania nie są niezależne i sprowadzają się do: F 2 = F 4 + F 5 (3) Liczba zmiennych NV = 11 (5 natężeń Ograniczenia dla ułamków masowych: przepływu, 6 ułamków masowych). x 11 + x 12 = 1 (4) Liczba równań NE = 7. x 31+ x 32 = 1 (5) x 41 +x 42 = 1 (6) Ograniczenia procesowe: F 5/F 4= 0, 585 (7) Liczba zmiennych decyzyjnych: ND = NV-NE= 11 7 = 4 Zgodnie z treścią zadania przyjmujemy: x 11, x 31, x 41 i F 1 jako zmienne decyzyjne (wiadome).

Przykład 2. Obliczyć skład roztworu otrzymanego przez rozpuszczenie w wodzie węglanu wapnia. Roztwór jest w kontakcie z atmosferą, ciśnienie cząstkowe CO 2= 30 Pa, T = 290 K. Rozwiązanie Zakładamy, że układ w stanie równowagi zawiera Ca. CO 3(s) oraz Ca 2+, CO 32 , HCO 3 , H+, OH i H 2 CO 3 (aq). W roztworze występuje sześć składników o nieznanych stężeniach, co wymaga sześciu niezależnych równań: pięć równań opisujących równowagi. równanie bilansu ładunków elektrycznych. Równanie bilansu materiałowego odrzucamy ze względu na możliwość pochłaniania lub wydalania CO 2 z układu.
 = Ca 2+ + CO 3")
Jedna z możliwych kombinacji: I. Ca. CO 3(s) = Ca 2+ + CO 3 2 [CO 32 ] [Ca 2+] = 2, 9 • 10 9 II. H 2 O(c) = H+ + OH [OH ] [H+] = 5, 5 • 10 15 III. H 2 CO 3(aq) = H+ + HCO 3 [HCO 3 ] [H+]/[H 2 CO 3] = 9, 3 • 10 8 IV. H 2 CO 3(aq) = H 2 O + CO 2 (g) [CO 2]/ [H 2 CO 3](aq) = 31, 7 V. HCO 3 = H+ + CO 32 [H+] [CO 32 ]/[HCO 3 ] = 1, 4 • 10 11 Bilans ładunków: VI. yi zawartość jonów „i” w 1 kg wody. Ponieważ zawartości jonów są bardzo małe, to przyjmujemy współczynniki aktywności i równe jedności. Ilość H 2 CO 3(aq) w 1000 g H 2 O można obliczyć z równania IV: dla p/po=30: 101325

y 1 - ilość jonów HCO 3 - w 1000 g wody, ilości pozostałych jonów w 1000 g wody można wyrazić równaniami: (z równania III) (z równania V) (z równania I) Podstawiając te wyrażenia do równania VI uzyskujemy: Układ 5 równań z 5 niewiadomymi – rozwiązać.
 Fe. O można zredukować do metalu mieszaniną 20% obj.")
Przykład 3 Tlenek żelaza (II) Fe. O można zredukować do metalu mieszaniną 20% obj. CO i 80% obj. N 2 pod ciśnieniem atmosferycznym i w T = 1270 K. Zakładając, że reakcja osiąga stan równowagi obliczyć masę żelaza otrzymanego przez przepuszczenie 1 m 3 gazu nad tlenkiem żelaza. Stała równowagi reakcji: Fe. O(s) + CO(g) Fe(s) + CO 2(g) w 1270 K ma wartość 0, 40. Suma ułamków molowych w stanie równowagi w fazie gazowe: Po podstawieniu: Przepuszczając 1 m 3 gazu nad Fe oczekuje się otrzymania: kg żelaza
 stwierdzono, że gazie wychodzącym jest 4%obj. CO 2.")
Po ustaleniu parametrów (T, p, GHSV) stwierdzono, że gazie wychodzącym jest 4%obj. CO 2. Obliczyć rzeczywistą ilość metalicznego Fe otrzymaną po przepuszczeniu 1 m 3 gazu przez Fe. O oraz termodynamiczną wydajność reakcji. Równowagowy stopień przereagowania CO wynosi: Rzeczywiście osiągnięty stopień przereagowania: Wydajność reakcji w stosowanych warunkach: Ilość otrzymanego metalicznego żelaza: 0, 142 • 0, 7 = 0, 099 kg. Termodynamiczna wydajność reakcji: A – substrat w niedomiarze stechiometrycznym

Przykład 4. Obliczyć równowagowe ciśnienie CO 2 powstającego w procesie dysocjacji termicznej Ca. CO 3 w 1000 K. log Kap, 1000 = 0, 9659 Kap, 1000 = 0, 1082 Dla reakcji: Ca. CO 3 Ca. O + CO 2

Przykład 5. Mieszaninę kwasu siarkowego i azotowego zawierającą odpowiednio 35 i 30% mas. tych kwasów (reszta woda) sporządza się ze: stężonego roztworu H 2 SO 4 o zawartości 95% mas. czystego składnika, roztworu HNO 3, w którym woda stanowi 35% mas. , mieszaniny kwasów z tzw. zawrotu, zawierającej 5% mas. H 2 SO 4 i 90% wody, resztę stanowi kwas azotowy. Sporządzić bilans urządzenia dla przygotowania 1200 kg/h produktu końcowego. F 3 x 32, x 33 F 2 F 1 x 21, x 22, x 23 F 4 x 41, x 42, x 43 x 11, x 13 1 kwas siarkowy, 2 kwas azotowy, 3 woda. Równania bilansowe: F 1 x 11 + F 2 x 21 = F 4 x 41 F 2 x 22 + F 3 x 32 = F 4 x 42 F 1 x 13 + F 2 x 23 + F 3 x 33 = F 4 x 43

Ograniczenia dla ułamków masowych: x 11 + x 13 = 1 x 21 + x 22 + x 23 = 1 x 32 + x 33 = 1 x 41 + x 42 + x 43 = 1 Ograniczenia procesowe brak. Liczba zmiennych NV = 14 Liczba niezależnych równań NE = 7 Liczba zmiennych decyzyjnych ND = NV – NE = 7 Podstawiamy: x 11 = 0, 95, x 21 = 0, 05, x 23 = 0, 90, x 33 = 0, 35, x 41 = 0, 35, x 42 = 0, 30, F 4 = 1200 kg/h Rozwiązanie: F 1 = 429, 76 kg/h, F 2 = 234, 42 kg/h, F 3 = 535, 82 kg/h, x 13 = 0, 05, x 22 = 0, 05, x 32 = 0, 65, x 43 = 0, 35 Do rozwiązania powyższego układu równań konieczna była znajomość 7 z 14 zmiennych. Nie mogą to być dowolne zmienne, lecz tylko taki zestaw, który zapewni niezależność równań w układzie równań.

Przykład 6. Dla reakcji dimeryzacji ditlenku azotu: 2 NO 2 N 2 O 4 stała równowagi Kap w 1000 K ma wartość: Kap = 0, 986 Obliczyć równowagowy stopień przemiany NO 2 w N 2 O 4 pod p = 0, 1 MPa oraz p = 1 MPa. W podanych warunkach ciśnienia Kap Kp. dla p = 0, 1 Mpa: Kx = Kp; dla p = 1 Mpa: bo = 1 Oznaczamy: strumień F 1 (wlotowy) NO 2: N 2 O 4 : x 11 = 1 x 12 = 0 strumień F 2 (wylotowy) x 21 = ? x 22 = ?

Układ równań do rozwiązania: pierwiastek N: F 1 = F 2 x 21+ 2 F 2 x 22 pierwiastek O: 2 F 1 = 2 F 2 x 21 + 4 F 2 x 21 + x 22 = 1 F 1 zmienna projektowa, przyjmujemy: 1 kmol • h 1 Rozwiązanie jest trywialne, ponieważ x 22 = 1 x 21 i ułamki molowe w stanie równowagi można obliczyć bez rozwiązywania równań bilansowych.

Przykład 7. Obliczyć równowagowy skład mieszaniny produktów reakcji dysocjacji termicznej pary wodnej w temperaturach 1273 i 2273 K przy ciśnieniu normalnym: 2 H 2 O(g) 2 H 2 + O 2 Stałe równowagi Kap: Kap (1273) = 2, 95 10 15 Kap (2273) = 3, 16 10 6 W podanych warunkach można założyć Kap = Kp. Ponieważ = 1 więc Kp = Ka tylko dla p = 0, 1 MPa. Ułamki molowe: strumień F 1 (wlotowy): H 2 O: x 11 = 1 H 2 : x 12 = 0 O 2 : x 13 = 0 strumień F 2 (wylotowy): x 21 = ? x 22 = ? x 23 = ? Układ równań do rozwiązania: pierwiastek H: F 1 = 1 kmol • h 1 = F 2 x 21 + F 2 x 22 pierwiastek O: F 1 = 1 kmol • h 1 = F 2 x 21 + 2 F 2 x 23 F 1, x 12, x 13 – zmienne projektowe
: SO")
Przykład 8. Gazy z rozkładu surowca pirytowego o składzie (w % obj. ): SO 2 = 7, 8%; O 2 = 10, 8%; N 2 = 81, 4% przesyła się do reaktora utleniania kontaktowego. W reaktorze utrzymywana jest temperatura 770 K, ciśnienie jest zbliżone do atmosferycznego. Obliczyć skład gazu opuszczającego reaktor przy założeniu, że reakcja osiąga stan równowagi. Stała równowagi reakcji: SO 2 + ½ O 2 = SO 3 wynosi Kap = Kp = 85 Wykonać bilans materiałowy. Oznaczenia ułamków molowych: strumień F 1 (wlot): SO 2 x 11 = 0, 078 O 2 x 12 = 0, 108 N 2 x 13 = 0, 814 SO 3 x 14 = 0 strumień F 2 (wylot): x 21 = ? x 22 = ? x 23 = ? x 24 = ? F 2 = ?

Układ równań do rozwiązania: S: F 1 x 11 = F 2 x 21 + F 2 x 24 O: 2 F 1(x 12 + x 11) = 2 F 2 x 21 + 3 F 2 x 24 + 2 F 2 x 22 N: 2 F 1 x 13 = 2 F 2 x 23 F 1 zmienna projektowa, jej wartość można założyć. Przyjmujemy F 1= 100 kmol • h 1. Układ równań można rozwiązać metodą numeryczną. Skład gazu na wylocie z reaktora: x 21=x*SO 2 = 0, 003; F 2 = 96, 28 kmol • h 1. x 22=x*O 2 = 0, 073; x 24=x*SO 3 = 0, 078; x. N 2 = 0, 846;
 kmol/h kg/h Rozchód")
Bilans materiałowy Składnik SO 2 N 2 SO 3 Przychód (substraty) kmol/h kg/h Rozchód (produkty) kmol/h kg/h F 1 • x 11 = 7, 80 499, 20 F 1 • x 12 = 10, 80 345, 60 F 1 • x 13 = 81, 40 2279, 20 F 1 • x 14 = 0, 0 F 2 • x 21 = 0, 29 18, 56 F 2 • x 22 = 7, 03 224, 96 F 2 • x 23 = 81, 45 2280, 60 F 2 • x 24 = 7, 51 600, 80 100 kmol/h 96, 28 kmol/h (3124, 92 kg/h) (3124, 0 kg/h)

Przykład 9. Określić ilość i równowagowy skład produktów zgazowania węgla brunatnego o składzie: c = 31, 55%mas. C, n = 0, 44% mas. N 2, h = 2, 56%mas. H 2, o = 8, 05% mas O 2 Czynnikiem zgazowującym jest woda zawarta w węglu. Obliczenia wykonać dla ciśnienia 0, 1 MPa i T = 950 K. W obliczeniach uwzględnić następujące reakcje: C + CO 2 = 2 CO Kp. B = 0, 6507 C + H 2 O = CO + H 2 Kp. W = 1, 1932 C + 2 H 2 = CH 4 Kp. M = 0, 1755 s = 0, 40% mas. S, w = 50, 0% mas. H 2 O,
 gazu powstającego")
Równania bilansowe dla 1 kg surowca węglowego: Oznaczamy: V ilość (m 3) gazu powstającego z 1 kg węgla, mc ilość moli nieprzereagowanego węgla pierwiastkowego, xi ułamek molowy składnika i, bilans węgla: bilans tlenu: bilans wodoru: Dodatkowo: suma ułamków molowych = 1 Podstawiamy: z 1 = x. H 2; z 2 = x. CO; z 3 = mc; z 4 = V;
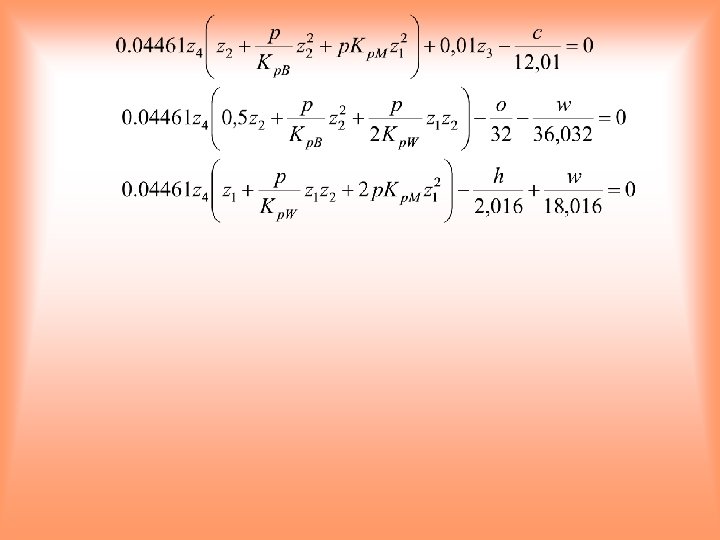

Przykład 10. Para wodna i metan w stosunku 5: 1 wchodzą do reaktora: T = 850 K, p = 0, 1 MPa. Obciążenie reaktora: 30 kmol CH 4/s. Obliczyć równowagowy skład gazów (poreakcyjnych). Ułożyć bilans materiałowy i cieplny reaktora. Uwzględnić następujące reakcje: CH 4 + H 2 O CO + 3 H 2 CO + H 2 O CO 2 + H 2 Kap 1 = 0, 574 W warunkach reakcji Kap Kp (1) (2) Ho(r 1)298 = 206250 J/mol Ho(r 2)298 = 41170 J/mol Kap 2 = 2, 21

x ilość moli CO, y ilość moli CO 2 Ilości moli reagentów w stanie równowagi dla reakcji (1): Ilości moli reagentów w stanie równowagi dla reakcji (2): Suma ilości moli n = 6 + 2 x. Ułamki molowe w stanie równowagi:

Zapisując stałe równowagi za pomocą ułamków molowych otrzymujemy dwa równania o dwóch niewiadomych: Rozwiązanie metodą kolejnych przybliżeń (kalkulator i wykres) daje skład mieszaniny w stanie równowagi.

1 – CH 4, 30 mol/h 2 – H 2 O, 150 mol/h 3 – CO 4 – H 2 5 – CO 2 F 3, x 31, x 32, x 33, x 34, x 35 Równania do rozwiązania: a) stałe równowagi: F 1 (H 2 O) b) równania bilansu materiałowego (na mol!): pierwiastek C: pierwiastek H: pierwiastek O: F 2 = F 3 x 31 + F 3 x 33 + F 3 x 35 F 1 + 2 F 2 = F 3 2 x 31 + F 3 x 32 + F 3 x 34 F 1 = F 3 x 32 + F 3 x 33 + F 3 2 x 35 c) ograniczenia dla ułamków molowych: x 31 + x 32 + x 33 + x 34 + x 35 = 1 F 2, (CH 4)

Bilans materiałowy Przychód Rozchód CH 4 H 2 O CO H 2 CO 2 30 150 0 481, 0 2702, 4 0 0 0 2, 3 103, 3 8, 0 100, 9 20, 2 180, 0 kmol • h 1 3183, 4 kg • h 1 234, 7 kmol • h 1 36, 9 1861, 0 224, 0 203, 4 888, 8 3214, 1 kg • h 1
: . . Zapotrzebowanie ciepła na przeprowadzenie")
Bilans cieplny Zapotrzebowanie ciepła na przeprowadzenie reakcji (1): . . Zapotrzebowanie ciepła na przeprowadzenie reakcji (2): . . Ilość ciepła potrzebnego do przebiegu reakcji (1) w temperaturze 850 K przy obciążeniu 30 mol CH 4 • s 1: x (obciążenie) x (stopień przereagowania w reakcji (1)) = = 223546 • 30 • 0, 92 = 6169, 9 k. J • s 1 Ilość ciepła wydzielającego się w reakcji (2) w tych samych warunkach: Q 2 = 36158 • 30 • 0, 65 = 705, 1 k. J • s 1 Zapotrzebowanie cieplne reaktora

Bilans cieplny reaktora: qi ciepło wnoszone przez 1 mol substancji „i”. Podobnie liczymy q. CO, q. CO 2, q. H 2.

Po podstawieniu otrzymanych wartości do równania bilansu otrzymujemy: 10849, 5 + Qdost = 9398, 3 + 5464, 8 + Qstrat Qdost =4180, 8 k. J • s 1 Zestawienie bilansu cieplnego procesu konwersji metanu. z metanem z wodą z tlenkiem węgla z ditlenkiem węgla z wodorem ciepło reakcji ciepło dostarczone ciepło strat przychód rozchód 28, 4 • 30= 852, 0 66, 65 • 150 = 9997, 5 0 0 0 705, 1 4180, 8 0 28, 4 • 2, 3 = 65, 3 66, 65 • 103, 3 = 6884, 9 18, 3 • 8, 0 = 146, 4 26, 03 • 20, 2 = 525, 8 17, 6 • 100, 9 = 1775, 8 6169, 9 0 0, 04 • Qdost = 167, 2 15735 k. J • s 1

Przykład 11. W palniku gaz generatorowy spalany jest z 20% owym nadmiarem O 2 z powietrza w stosunku do ilości tlenu niezbędnej do całkowitego spalania: Skład gazu (% obj. ): CO: 28, 0; CO 2: 3, 5; O 2: 0, 5; N 2: 68, 0 Obliczyć skład i strumień masowy produktów spalania, jeżeli do palnika dopływa 100 kmol*h gazu generatorowego i z tego 98% ulega spaleniu. F 1, x 12, x 13, x 14 F 3, x 31, x 32, x 33, x 34 F 2, x 23, x 24 1 – CO; 2 – CO 2; 3 – O 2; 4 – N 2 CO + ½ O 2 CO 2 = 98%

Aby ułożyć równanie bilansowe, należy wpierw obliczyć wejściowy strumień powietrza. Bilans tlenu (kmol h 1): - tlen niezbędny do spalenia CO: 28, 0 • ½ = 14 - tlen w gazie generatorowym = 0, 5 - zapotrzebowanie tlenu netto: 14 0, 5 =13, 5 - tlen z powietrza: 13, 5 • 1, 2 = 16, 2 - tlen rzeczywiście zużyty: 0, 98 • 16, 2 = 15, 9 - tlen w produktach reakcji: 16, 2 + 0, 5 – 15, 9 = 0, 8 Azot dopływający w strumieniu powietrza 79/21 • 16, 2 = 60, 9. Charakterystyka strumieni wejściowych: F 1 = 100 kmol • h 1 x 11 = x. CO = 0, 28 x 12 = x. CO 2 = 0, 035 x 13 = x. O 2 = 0, 005 x 14 = x. N 2 = 0, 68 F 2 = 16, 2 + 60, 9 = 77, 1 kmol • h 1 x 23 = x. O 2 = 0, 21 x 24 = x. N 2 = 0, 79

Układ równań bilansowych: C: F 1 x 11 + F 1 x 12 = 28, 0 + 3, 5 = 31, 5 = F 3 x 31 + F 3 x 32 O: ½ F 1 x 11 + F 1 x 12 + F 1 x 13 + F 2 x 23 = 34, 2 = ½ F 3 x 31 + F 3 x 32 + F 3 x 33 N: F 1 x 14 + F 2 x 24 = 68 + 60, 9 = 128, 9 = F 3 x 34 Równanie określające stopień przereagowania w stosunku do reagenta limitującego: Zastępując Fixij molowym natężeniem przepływu fij otrzymujemy układ równań liniowych: f 31 + f 32 = 31, 5 ½ f 31 + f 32 = f 33 = 34, 2 f 34 = 128, 9 f 31 + f 32 + f 33 + f 34 = F 3 (28 – f 31)/28 = 0, 98 Rozwiązanie daje wartości molowych natężeń przepływu zestawienie bilansowe: przychód – rozchód.

Efekt cieplny reakcji

Przykład 1 Znając standardowe wartości entalpii tworzenia NO, N 2 O i H 2 O, obliczyć standardową entalpię reakcji: 2 NO + H 2 = N 2 O + H 2 O w temperaturze 298 K. Hotw. NO, 298 = 90, 4 k. J • mol 1, Hotw. N 2 O, 298 = 81, 5 k. J • mol 1, Hotw. H 2 O, 298 = 285, 8 k. J • mol 1, Zgodnie z prawem Hessa standardową entalpię reakcji w 298 K można obliczyć z zależności: Standardowa entalpia reakcji redukcji NO wodorem do N 2 O wynosi 384, 7 k. J • mol 1.
: 2 Na. HCO 3(s) =")
Przykład 2 Obliczyć wartość entalpii reakcji (w 383 K): 2 Na. HCO 3(s) = Na 2 CO 3(s) + CO 2(g) + H 2 O(g) Zadanie można rozwiązać dwoma metodami: 1. Korzystając z danych opisujących zależność entalpii poszczególnych reagentów od temperatury. 2. Obliczając Hor, 298 (prawo Hessa), a następnie Cp i Hor, 383 (prawo Kirchhoffa). Ad 1. Potrzebna jest znajomość zależności entalpii od temperatury: Hi = f(T). Wówczas: dla kalcynacji wodorowęglanu sodu na podstawie danych tablicowych: Dla T = 383 K, Hor, 383 = 128, 64 k. J • mol 1.
: Podstawiając tablicowe wartości")
Ad 2. Obliczenie entalpii reakcji w temperaturze 298 K (prawo Hessa): Podstawiając tablicowe wartości Hotw. i, 298 uzyskujemy Hor, 298 = 128, 518 k. J • mol 1. Dla obliczenia Hor, 383 korzystamy z prawa Kirchhoffa: T = 383 K Potrzebna jest znajomość zależności Cp = f(T) tablice. Po podstawieniu otrzymujemy Hor, 383 = 128, 61 k. J • mol 1 (różnica < 0, 03%). Dla poszczególnych substancji w literaturze prezentowane są niekiedy dane w postaci funkcji (HT H 298) = f(T) oraz Hotw. i = f(T). Takie dane znacznie upraszczają obliczenia Hor.

Przykład 3 Obliczyć standardową entalpię reakcji tworzenia amoniaku w 600 K: ½ N 2 + 3/2 H 2 = NH 3 Dane: Hor, 298 = 46, 26 k. J • mol 1 (H 600 H 298)N 2 = 8, 901 k. J • mol 1 (H 600 H 298)H 2 = 8, 813 k. J • mol 1 (H 600 H 298)NH 3 = 12, 079 k. J • mol 1 Entalpia reakcji w temperaturze T wynosi: ponieważ: więc:

Zadanie można także rozwiązać na podstawie zależności: Hotw, NH 3, 600 = 51, 75 k. J • mol 1 Obliczone wartości entalpii tworzenia NH 3 w 600 K różnią się o 0, 2% i są zgodne z wartością Hotw, NH 3 obliczoną z zależności temperaturowych Cp ( 51, 8 k. J • mol 1).

Przykład 4 Obliczyć entalpię krakowania propanu na eten i metan w 753 K i pod p = 6, 89 Mpa. C 3 H 8 = CH 4 + C 2 H 4 Obliczenie entalpii reakcji w podanych warunkach wykonujemy w 4 etapach: 1. Obliczenie entalpii standardowej reakcji w temperaturze 298 K. 2. Obliczenie entalpii reakcji w 753 K i pod ciśnieniem 0, 1013 MPa. 3. Obliczenie zmiany entalpii reakcji spowodowanej zmianą ciśnienia od 0, 1013 MPa do 6, 89 MPa. 4. Obliczenie entalpii reakcji w temperaturze 753 K i pod ciśnieniem 6, 89 MPa,

Ad 1. Hotw, 298, C 3 H 8 = 103, 90 k. J • mol 1 Hotw, 298, C 2 H 4 = 52, 32 k. J • mol 1 Hotw, 298, CH 4 = 74, 88 k. J • mol 1 Hor, 298 = Hor, 298, CH 4 + Hor, 298, C 2 H 4 Hor, 298, C 3 H 8 = 81, 34 k. J • mol 1 Hor, 298 można także obliczyć na podstawie wartości ciepła spalania produktów i surowców: Qos, C 3 H 8 = 2220, 4 k. J • mol 1 Qos, C 2 H 4 = 1411, 6 k. J • mol 1 Qos, CH 4 = 890, 6 k. J • mol 1 Hor, 298 = Qos, C 3 H 8 Qos, C 2 H 4 Qos, CH 4 = 81, 8 k. J • mol 1 Do dalszych obliczeń bierzemy wartość uzyskaną pierwszym sposobem.
![Ad 2. Zależność cp od temperatury: [J/mol] Stąd: Entalpię reakcji w temperaturze 753 K](http://slidetodoc.com/presentation_image_h/0474a12ecd2443024868c0668d6f15c8/image-41.jpg "Ad 2. Zależność cp od temperatury: [J/mol] Stąd: Entalpię reakcji w temperaturze 753 K")
Ad 2. Zależność cp od temperatury: [J/mol] Stąd: Entalpię reakcji w temperaturze 753 K obliczamy z prawa Kirchhoffa:

Ad 3. Zmianę entalpii reakcji spowodowaną wzrostem ciśnienia od 0, 1013 MPa do 6, 89 MPa w temperaturze 753 K obliczamy stosując wykres Hougena Watsona. Niezbędne wartości parametrów krytycznych znajdujemy w tablicach i na wykresach w zmiennych zredukowanych. Ad 4. Obliczamy wartość Hor, 753, p: Entalpia krakowania propanu w 753 K i pod p = 6, 89 MPa wynosi 82, 9 k. J • mol 1. Ciśnienie wywiera istotnego wpływu na wartość entalpii tej reakcji, jego wpływ można pominąć.

Przykład 5 Obliczyć przybliżoną wartość entalpii reakcji kalcynacji wodorowęglanu sodu znając dwie wartości prężności gazowych produktów kalcynacji, pozostających w stanie równowagi z fazą stałą. T 1 = 303 K p 1 = 0, 827 k. Pa, T 2 = 383 K p 2 = 167, 0 k. Pa. Kalcynacja: 2 Na. HCO 3(s) = Na 2 CO 3(s) + CO 2(g) + H 2 O(g)

Zakładamy, że entalpia tej reakcji jest stała w rozpatrywanym zakresie temperatury. Możemy ją obliczyć korzystając z równania izobary Van't Hoffa: Całkując to wyrażenie otrzymujemy: lub: (A = Hor/2, 303 R) Równanie reakcji: 2 Na. HCO 3(s) = Na 2 CO 3(s) + CO 2(g) + H 2 O(g) Stała równowagi tej reakcji: Aktywności Na. HCO 3 i Na 2 CO 3 są równe jedności, a zamiast aktywności H 2 O i CO 2 można podstawić wartości ich prężności (niskie ciśnienie). Uzyskujemy: Z równania reakcji wynika, że prężności H 2 O i CO 2 są równe, więc:

Z wartości prężności produktów gazowych można zatem obliczyć wartości stałych równowagi w obu temperaturach: z układu równań: wyznaczamy wartość stałej A: A = 6681 Hor = A • 2, 303 R = 6681 • 2, 303 • 8, 314 = 127, 9 k. J • mol 1 Entalpia tej reakcji obliczona z danych termochemicznych wynosi 128, 6 k. J • mol 1. Błąd obliczenia z zastosowaniem danych równowagowych wynosi 0, 5%.

Przykład 6 Określić zależność entalpii reakcji spalania wodoru w tlenie od temperatury oraz wyznaczyć ciepło tej reakcji w 500 K. H 2(g) + ½ O 2(g) = H 2 O(g) Dane: zależność stałej równowagi reakcji (tabela) oraz ciepeł molowych reagentów od temperatury. Temperatura, K log Kp 1000 1200 1400 1500 1750 2000 10, 059 7, 893 6, 340 5, 716 4, 365 3, 528

Zależność entalpii reakcji od temperatury jest określona prawem Kirchhoffa: JH stała całkowania. Dla wyznaczenia Hor w funkcji temperatury obliczany Cp: Cp = 15, 00 + 7, 87 • 10 3 T + 3, 93 • 105 T 2 a następnie obliczamy Hor, T: (1) Ponieważ brakuje danych umożliwiających obliczenie JH zależność ciepła reakcji od temperatury określamy metodą graficzną, korzystając z zależności stałej równowagi od temperatury (izobara Van’t Hoffa): Za Hor podstawiamy określoną uprzednio prawą stronę wyrażenia (1) i całkujemy: Jk stała całkowania

Dla wyznaczenia stałych całkowania JH i Jk przekształcamy uzyskane równanie: Prawą stronę równania oznaczamy symbolem J i obliczamy jej wartości dla temperatury, dla której znane są wartości stałej Kp. Robimy wykres zależności J = f(1/T) tg = JH, punkt przecięcia z osią rzednych (ekstrapolacja) = Jk Odczytana z wykresu wartość Jk = 56, 2, wartość stałej całkowania JH = 236100 J. Zależność ciepła reakcji spalania wodoru w tlenie od temperatury: Efekt cieplny tej reakcji w 500 K, obliczony według otrzymanego równania, wynosi: 234, 4 k. J • mol 1.

Przykład 7 Obliczyć dolną wartość opałową węgla o składzie 67, 9% C, 1, 5% H, 1, 6% O, 1, 0% N, 1, 3% S, 6, 5% wilgoci (reszta popiół). Ilość ciepła uzyskaną podczas spalania paliw nazywa się wartością opałową. Wyróżnia się dolną i górną wartość opałową. Dolna wartość opałowa zakładamy, że woda pozostaje w spalinach w postaci pary. Górna wartości opałowa zakładamy całkowite wykroplenie wody ze spalin. Doświadczalne wartości opałowe są w tablicach lub wyznacza się je eksperymentalnie. Można oszacować dolną wartość opałową ze wzoru Mendelejewa Qd dolna wartość opałowa, kcal • mol 1 C, H, O, S, W zawartość procentowe węgla, wodoru, tlenu, siarki i wilgoci w paliwie, Korzystamy ze wzoru Mendelejewa: Qd = 5822 kcal • mol 1 = 24 370 k. J • mol 1

Przykład 8 Obliczyć teoretyczną temperaturę spalania CO bez nadmiaru powietrza zakładając, że substraty mają temperaturę 298 K i znajdują się pod p = 1, 013 • 105 Pa. Dane: 1 cal = 4, 1868 J Dla uzyskania rozwiązania korzystamy z równania bilansu cieplnego: Bilans cieplny obliczamy dla 1 mola CO zgodnie z równaniem reakcji: CO + ½ O 2 + ½ • 79/21 N 2 = CO 2 + 79/42 N 2

Korzystając z wartości entalpii tworzenia CO i CO 2 obliczamy Hor, 298: Traktując azot jako produkt, który nie bierze udziału w reakcji Hprod obliczamy z: Temperatura substratów wynosiła 298 K, więc Hsub = 0. Równanie bilansu cieplnego przyjmuje postać: Obliczona teoretyczna temperatura spalania CO wynosi 2670 K.

Przykład 9 Obliczyć teoretyczną temperaturę spalania metanu ze stechiometryczną ilością powietrza. Temperatura reagentów wynosi 298 K, podczas spalania ciśnienie zachowuje stałą wartość, a spalaniu ulega tylko 80% metanu. Ciepła molowe: Ciepła tworzenia i parowania:

Rozwiązanie dwa sposoby. 1. Na entalpię wody w stanie gazowym w temperaturze reakcji składa się: entalpia H 2 O(c) wynikająca ze wzrostu temperatury od 298 do 373 K, entalpia parowania H 2 O w 373 K, entalpia pary H 2 O związana ze wzrostem temperatury od 373 K do T. Do obliczeń Hor, 298 w tej metodzie stosujemy Hotw, H 2 O(c) 298. 2. Entalpia wody jest równa entalpii pary wodnej z uwzględnieniem wzrostu jej temperatury od 298 K do T. Do obliczenia Hor, 298 stosujemy Hotw, H 2 O(g) 298. Równanie reakcji: CH 4 + 2 O 2 + 7, 52 N 2 = CO 2 + 2 H 2 O + 7, 52 N 2 Ogólne równanie bilansu cieplnego procesu ulega uproszczeniu (gdyż Hsub = 0 ):
. Wzór substancji CH")
Bilans materiałowy procesu dla 1 mola CH 4 (80% przereagowania substratów). Wzór substancji CH 4 O 2 N 2 CO 2 H 2 O Liczba moli substratów w mieszaninie substratów 1, 00 2, 00 7, 52 0, 00 Liczba moli substancji w mieszaninie produktów 0, 20 0, 40 7, 52 0, 80 1, 60 Ad. 1. Podstawiając uzyskane wyrażenie zamiast Hprod i wartość liczbową Hor, 298 do równania bilansu cieplnego otrzymujemy: T = 2034 K
: Podstawiamy uzyskane wyrażenie")
Ad 2. Wyrażenie na Hprod jest inne niż w wariancie (1): Podstawiamy uzyskane wyrażenie określające Hprod oraz wyliczoną wartość liczbową Hor, 298 do równania bilansowego otrzymujemy: 0, 00698 T 2 + 72, 18 T 175537 = 0 T = 2034 K Wyniki obliczeń według obydwu sposobów są identyczne. Zadowalającą dokładność obliczeń można uzyskać stosując uproszczony sposób (2) pod warunkiem użycia odpowiedniej wartości Hotw, H 2 O(g).

Przykład 10 Obliczyć teoretyczną temperaturę reakcji tlenku azotu ze stechiometryczną ilością chloru: NO + ½ Cl 2 = NOCI Temperatura substratów 298 K, założyć całkowite przereagowanie. Dane Hotw, NO, 298 = 90, 37 k. J • mol 1 Hotw, NOCl, 298 = 52, 59 k. J • mol 1 Ponieważ Hsub = 0 i jest tylko jeden produkt reakcji więc równanie bilansu cieplnego ma postać: Obliczamy wartość Hor, 298:
")
Podstawiamy do równania bilansowego wartość Hor, 298 oraz wyrażenie na (HT –H 298) i rozwiązujemy równanie: T = 1081 K Teoretyczna temperatura reakcji wynosi 1081 K. Rzeczywista temperatura reakcji jest znacznie niższa ze względu na fakt, że stopień przereagowania reagentów jest niższy od jedności.

Przykład 11 Wyznaczyć temperaturę końcową reakcji neutralizacji 200 kg 20% owego r ru H 2 SO 4 za pomocą 300 kg 12% owego r ru Na. OH. Temperatura reagentów 298 K, a wielkość strat ciepła do otaczającego środowiska jest równa 10% ciepła reakcji. Ho. H 2 SO 4, 298 = 210800 kcal • kmol 1 Ho. Na 2 SO 4, 298 = 328100 kcal • kmol 1 Ho. H 2 O, 298 = 68370 kcal • kmol 1 Ho. Na. OH, 298 = 112700 kcal • kmol 1 Obliczany początkowe liczby moli reagentów: wi, Gi, Mi odpowiednio: ułamek masowy składnika, masa roztworu składnika i masa molowa składnika i.

Ponieważ reakcja zachodzi wg równania: H 2 SO 4 + 2 Na. OH = Na 2 SO 4 + 2 H 2 O więc powstało 0, 408 kmol Na 2 SO 4 i pozostało 0, 084 kmol nieprzereagowanego Na. OH. Efekt cieplny neutralizacji: Straty ciepła wynoszą 10% ciepła reakcji ciepło powodujące podgrzanie reagentów: Temperatura końcowa roztworu: G masa roztworu, cm ciepło właściwe, Tk temperatura końcowa produktu, Tp temperatura surowców

Ciepło właściwe roztworu obliczamy sumując iloczyny ciepeł właściwych i udziałów masowych poszczególnych składników zgodnie z równaniem: Ciepła właściwe (kcal • kg 1 • K 1): c. Na 2 SO 4 =0, 231 c. H 2 SO 4 =0, 355 c. H 2 O =1, 000 Po podstawieniu: temperatura końcowa reakcji = 321, 4 K. Podstawą obliczeń teoretycznej i rzeczywistej temperatury proces było prawidłowe ułożenie bilansu cieplnego.

Przykład 12 Obliczyć temperaturę, z jaką mieszanina 11% obj. NH 3 z powietrzem powinna być wprowadzona do reaktora, aby utlenianie NH 3 przebiegało autotermicznie w 1100 K. Założyć, że straty ciepła stanowią 4% ciepła dostarczonego w procesie wstępnego ogrzewania surowców od T = 298 K. Dla rozwiązania zastosujemy bilans ciepła wykorzystujący Hor, w temperaturze procesu. Wychodzimy z ogólnego równania bilansu cieplnego: Zapisujemy to równanie w postaci: Tk temperatura końcowa procesu (produktów), Tp temperatura początkowa substratów. Hor, 298 obliczamy z równania:

Po podstawieniu i uproszczeniu: Na podstawie treści zadania sporządzamy schemat: Q 1 Q 2 Hr, 1100 (substraty)298 (substraty)Tp (substraty)1100 (produkty) 1100 Równanie bilansu cieplnego z uwzględnieniem strat cieplnych ma postać: (1) zależności ciepła molowego i ciepła reakcji od temperatury:
 po rozpisaniu: Po podstawieniu wartości liczbowych: Tp ~362 K")
Równanie (1) po rozpisaniu: Po podstawieniu wartości liczbowych: Tp ~362 K

Przykład 13 Mieszaninę gazów: 14% obj. SO 2, 7% obj. O 2 i 79% obj. N 2, otrzymaną w procesie prażenia pirytu, poddano utlenianiu na katalizatorze. Zakładając adiabatyczny przebieg procesu oszacować temperaturę katalizatora, jeżeli temperatura wprowadzonej mieszaniny gazów wynosi 803 K (ciśnienie 0, 1 MPa). Wariant 1. Bardzo uproszczony. Zakładamy, że reakcja utleniania zachodzi do końca. Wariant 2. Zakładamy, że na katalizatorze ustala się stan równowagi. Dane: zależność temperaturowa stałej równowagi reakcji tworzenia tritlenku siarki (tablica).

1. Wariant uproszczony Obliczenia wykonano dla takiej ilości gazu, które zawiera 1 mol SO 2. Otrzymuje się: a) ilość ciepła potrzebna do ogrzania mieszaniny reagentów do temperatury T: b) ciepło reakcji w temperaturze T wynosi Warunek adiabatyczności procesu: Rozwiązując to równanie otrzymuje się wartość T ~1160 K.

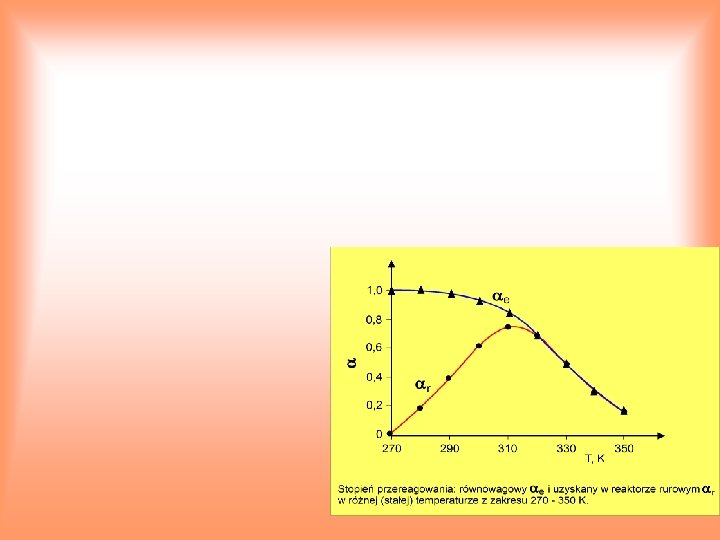
2. Wariant dokładny Oznaczając przez równowagowy stopień przereagowania SO 2 otrzymujemy związek pomiędzy stałą równowagi Kp oraz : SO 2 + ½ O 2 = SO 3 (1 ) (0, 5 ) 7, 15 0, 5 sumaryczna liczbą moli mieszaniny reagentów, Kp jest funkcją temperatury, więc stopień przereagowania zależy od temperatury. Rozwiązanie zmodyfikowanego równania bilansu cieplnego: można uzyskać metodą graficzną. Jako wynik rozwiązania otrzymuje się wartości: T = 970 K, = 0, 35. Temperatura według wariantu dokładnego jest niższa niż wg wariantu uproszczonego. Rzeczywista temperatura procesu będzie jeszcze niższa z powodu nieuniknionych strat cieplnych oraz nieosiągania stanu równowagi (skończony czas reakcji).

KINETYKA

Przykład 1 W reakcji pierwszego rzędu w ciągu 17 min przereagowało 50% substratu. Obliczyć, po jakim czasie przereaguje 90% substratu a po jakim 99%. A P co, A stężenie początkowe substratu A x – stężenie chwilowe produktu Dla reakcji pierwszego rzędu: Stała całkowania – z warunków początkowych: t=0 x = 0 Czas połowicznej przemiany – czas, po którym stężenie substratu maleje do połowy. Dla reakcji pierwszego rzędu podstawiając c. A = ½ co, A:


Przykład 2 W reakcji drugiego rzędu pomiędzy dwoma substratami o jednakowych stężeniach początkowych, w ciągu 500 s przereagowało 20% substratu. Obliczyć, po jakim czasie przereaguje 50% substratów. A + B P lub a, b stężenie początkowe substratu A i B x – stężenie chwilowe produktu Dla a = b oraz biorąc pod uwagę warunki początkowe ( t= 0 x = 0) (1)
 Dzieląc")
Dla c. A = ½ co, A można wprowadzić okres połowicznej przemiany: (2) Dzieląc (2) przez (1) otrzymujemy: skąd Jeżeli przereaguje 20% substratów to x = 0, 2 a; a x = 0, 8 a, zatem:

Przykład 3 W reaktorze rurowym o średnicy 2 cm i długości 40 cm badano kinetykę drugorzędowej reakcji zmydlania octanu etylu ługiem sodowym w roztworze wodnym (T = 323 K). U wlotu do reaktora bardzo szybko mieszano r r octanu etylu z ługiem i otrzymany r r, w którym stężenia obu substratów wynosiły 0, 030 mol/dm 3, tłoczono przez reaktor z szybkością 0, 5 cm 3/s. Stężenie jonów OH na wylocie z reaktora wynosiło 0, 0080 mol/dm 3. Obliczyć stałą szybkości reakcji. CH 3 COOC 2 H 5 + OH = CH 3 COO + C 2 H 5 OH Zmydlanie estru jest reakcją odwracalną, ze względu na dużą wartość stałej równowagi można przyjąć, że cały ester ulega hydrolizie. Reakcja drugiego rzędu, takie same stężenia początkowe obu substratów, więc można skorzystać z całkowej postaci równania kinetycznego: Zamiast t użyjemy czasu pobytu v: x = 8 10 3 mol dm 3 a = 30 10 3 mol dm 3 k = 0, 0482 mol 1 dm 3 s 1

Przykład 4 Na reakcję chemiczną składają się trzy stadia elementarne: A + B C + B (1) C + B A + B (2) C D (3) Podać wyrażenie określające szybkość tworzenia produktu D w warunkach przybliżenia stanu stacjonarnego. Jaka jest postać równania szybkości przy założeniu, że stadium (2) lub stadium (3) decyduje o szybkości reakcji. Przyrównując do zera szybkość zmiany stężenia produktu pośredniego otrzymujemy: Zatem w przybliżeniu stanu stacjonarnego szybkość tworzenia produktu D wynosi:
 jest etapem dużo wolniejszym od")
Dalsze uproszczenie tego wyrażenia można otrzymać gdy: stadium (3) jest etapem dużo wolniejszym od reakcji (2), tzn. gdy k 2 c. Bc. C >> k 3 c. C (czyli k 2 c. B >> k 3), wówczas: w przeciwnym przypadku, tzn. gdy k 2 c. B << k 3

Przykład 5 W reakcji otrzymywania tritlenku siarki na powierzchni katalizatora zachodzą reakcje: 2 X + O 2 = 2 XO XO + SO 2 = SO 3 + X (1) (2) Pomijamy desorpcję zaadsorbowanego SO 3 Wyprowadzić wyrażenie określające szybkość procesu w warunkach przybliżenia stanu stacjonarnego. Rozważyć przypadek, gdy reakcja (1) limituje szybkość procesu, a reakcja (2) osiąga stan równowagi. W warunkach stanu stacjonarnego szybkość powstawania pośredniego XO na centrum aktywnym X w reakcji (1) jest równa szybkości jego zanikania w reakcji (2): (1) Zakładając, że suma stężenia centrów aktywnych jest równa stężeniu katalizatora: (2) z obu równań można wyznaczyć stężenia centrów aktywnych. Jednak uzyskane w ten sposób wyrażenie określające szybkość reakcji: 2 SO 2 + O 2 = 2 SO 3 jest bardzo skomplikowane.
 limituje szybkość procesu, a stadium (2) jest w równowadze, tzn:")
Przyjmując, że stadium (1) limituje szybkość procesu, a stadium (2) jest w równowadze, tzn: to warunek ten łącznie z równaniem (2) pozwala rozpisać stężenia c. X i c. XO pomocą stężeń trwałych połączeń i sprowadzić równanie szybkości do postaci: Stężenie centrów aktywnych na powierzchni danego katalizatora ckat jest stałe. Nie zmniejszając ogólności wyrażenia można przyjąć ckat = 1 i przekształcić wyrażenie określające r:

Przykład 6 W układzie heterogenicznym: ciało stałe – gaz, w warstwie międzyfazowej zachodzi reakcja pierwszego rzędu. Reagent A, ze strumienia gazu o stężeniu c. A, dyfunduje do powierzchni między fazowej i osiąga tam stężenie c. AS. Szybkość reakcji chemicznej na granicy faz jest funkcją stężenia c. AS. Napisać wyrażenie określające szybkość przemiany w warunkach stanu ustalonego. W stanie ustalonym, ilość substratu A doprowadzanego do powierzchni w drodze dyfuzji jest równa ilości substratu ulegającej przereagowaniu w jednostce czasu i na jednostkę powierzchni rozdziału faz: k. C współczynnik wnikania masy, DA współczynnik dyfuzji substancji A, x grubość warstwy laminarnej na powierzchni stałego reagenta, k. S stała szybkości reakcji powierzchniowej,

Z równości wyznaczymy stężenie reagenta na powierzchni międzyfazowej: Podstawiając za c. AS otrzymujemy: Stałą szybkości przemiany (k) określa wyrażenie: Opór przemiany jest równy sumie oporu kinetycznego 1/k. S i dyfuzyjnego 1/k. C. Jeżeli wartość stałej szybkości reakcji chemicznej znacznie przewyższa wartość współczynnika wnikania masy, wyrażenie na szybkość upraszcza się do: tzn. opór dyfuzji określa szybkość przemiany proces przebiega w obszarze dyfuzyjnym. W przeciwnym przypadku, (tj. gdy k. C >> k. S): Stężenie reagenta w objętości i na powierzchni reakcji jest w przybliżeniu jednakowe, a dyfuzja nie wpływa na szybkość przemiany – kinetyczny obszar przebiegu reakcji. Uwaga: Sumowanie oporów poszczególnych etapów jest możliwe tylko w przypadku liniowej zależności szybkości tych etapów od siły napędowej (reakcji, dyfuzji).

Przykład 7 Badając reakcję katalitycznej izomeryzacji cis 2 butenu w 300 K, otrzymano zestaw wartości stężeń cis butenu 2 (A) i trans butenu 2 (B) w czasie. Oszacować wartości stałych szybkości reakcji w badanym układzie przyjmuąc, że wszystkie stadia są reakcjami pierwszego rzędu. Przemiana zachodzi w układzie trójskładnikowym, który można opisać schematem: buten 1 (C) cis buten 2 (A) Model kinetyczny procesu: trans buten 2 (B) Wartości stałych równowagi otrzymuje się ze stężeń zmierzonych po nieskończenie długim czasie reakcji.

Przykłady obliczeń projektowych

Przykład 1. Do reaktora przepływowego o objętości V = 10 dm 3 zaopatrzonego w mieszadło doprowadza się wodny roztwór bezwodnika kwasu octowego o strumieniu objętościowym q = 0, 04 dm 3 s-1 i stężeniu c 0 = 300 mol m-3. Stała szybkości reakcji: k (CH 3 CO)2 O + H 2 O 2 CH 3 COOH (V) prowadzonej w dużym nadmiarze wody wynosi k = 0, 0051 s-1. Obliczyć stężenie bezwodnika oraz kwasu w strumieniu opuszczającym reaktor w stanie ustalonym.

Rozwiązanie c. A - stężenie bezwodnika kwasu c. B - stężenie kwasu Z dobrym przybliżeniem spełniona jest równość: (55) Równanie ciągłości składnika A w stanie ustalonym ma postać: (56) można z niej obliczyć stężenie substratu A w strumieniu wylotowym z reaktora: (57) = V/q - umowny czas przebywania (w warunkach zadania 250 s) Z (57) otrzymujemy końcowe stężenie bezwodnika: Stopień przereagowania substratu A wynosi: Stężenie kwasu octowego wynosi (dla (55)) 336, 2 mol m-3
, to stopień przereagowania wynosiłby")
Jeżeli umowny czas przebywania byłby dwukrotnie większy (tj. 500 s), to stopień przereagowania wynosiłby ~0, 72. Dalsze zwiększanie czasu przebywania powoduje coraz mniejszy przyrost stopnia przereagowania. Dla umownego czasu przebywania = 1000 s, stopień przereagowania osiągnie wartość ~0, 84.

Przykład 2 W pojedynczym reaktorze przepływowym z doskonałym mieszaniem w reakcji nieodwracalnej pierwszego rzędu osiąga się stopień przereagowania równy 0, 9. Jeśliby zastąpić ten reaktor baterią dwóch bliźniaczych reaktorów połączonych ze sobą szeregowo, to: • jaki stopień przereagowania można uzyskać przy takiej samej objętości baterii jaką miał reaktor pojedynczy, jeżeli zachowa się również strumień objętości (przypadek A)? • o ile zwiększy się strumień objętości, jeżeli zachowa się taką samą przemianę substratu (przypadek B)?
 Przypadek A -")
Rozwiązanie Dla pojedynczego reaktora z równania ciągłości substratu otrzymuje się: (58) Przypadek A - zachowanie tego samego czasu przebywania Ponieważ w baterii reaktorów czas przebywania b = p, więc dla takich samych objętości obu reaktorów baterii, czas przebywania w każdym z tych reaktorów wyniesie = 4, 5 jednostek czasu (k p = 9 dla baterii dwóch reaktorów, dla pojedynczego reaktora = 4, 5). W pierwszym reaktorze stężenie substratu w strumieniu opuszczającym reaktor wyniesie: (59)
 Z zależności (59) i")
W drugim reaktorze nastąpi dalsze zmniejszenie stężenia do wartości: (60) Z zależności (59) i (60) otrzymujemy: (61) gdzie: Stopień przereagowania obliczamy z równania: W baterii reaktorów, o objętości takiej samej jak pojedynczy reaktor z doskonałym mieszaniem, stopień przereagowania jest większy; w warunkach przykładu większy o 0, 067.

Przypadek B - zachowanie tego samego stopnia przereagowania Jeżeli stopień przereagowania w baterii będzie taki sam jak w pojedynczym reaktorze, to z równań (59) i (60) otrzymujemy: (60) oczywiście 1 = 2 Wielkość k 1 jest więc równa: Stosunek wielkości k p odpowiadający pojedynczemu reaktorowi i wielkości (k 1)b odpowiadającej baterii (objętości reaktorów zestawionych w baterii są sobie równe) - jest równy stosunkowi strumieni objętości: baterii i pojedynczego reaktora: (61) Strumień objętościowy reagentów w baterii reaktorów z doskonałym mieszaniem można zwiększyć 2, 1 raza w stosunku do strumienia objętości dla pojedynczego reaktora z doskonałym mieszaniem.

Przykład 3 Wodną zawiesinę krochmalu poddaje się hydrolizie kwasowej w reaktorze zbiornikowym o pracy okresowej według sumarycznego równania reakcji: k k (C 6 H 1005)n + (n - 1)H 20 . . . n C 6 H 12 O 6 (VI) Z zawiesiny poreakcyjnej można w dalszej przeróbce wydzielić wytworzony cukier gronowy. - Podać zależność określającą stopień rozkładu krochmalu w czasie reakcji w objętości V. - Obliczyć średnią produkcję cukru na godzinę, jeżeli w jednej szarży poddaje się hydrolizie 200 kg krochmalu w przeliczeniu na czysty substrat. Założyć, że czas napełniania i opróżniania reaktora wynosi 1400 s, a całkowity czas całego cyklu - 10 800 s. Stała szybkości reakcji wynosi k= 0, 0001 s-1. Zaniedbać reakcję zachodzącą w okresie napełniania i opróżniania reaktora.

Rozwiązanie A - stężenie krochmalu. Dla reakcji pierwszego rzędu, stopień przereagowania substratu A wynosi: = 1 - exp(-kt) = 1 - exp(- 0, 94) = 0, 609 (t = tc-tj) Wartość stopnia przereagowania nie zależy od początkowego stężenia substratu oraz od objętości reaktora V. Zgodnie z równaniem stechiometrycznym z 1 kg substratu A otrzymuje się ~1, 11 kg cukru gronowego B. Skoro stopień przereagowania wynosi , to z 1 kg A otrzyma się: m. B = 1, 11 kg B z 1 kg A. Średnią produkcję (na godzinę) można obliczyć, uwzględniając rzeczywisty czas reakcji i czas biegu jałowego reaktora: Produkcja w jednej szarży wynosi 135 kg 3 h). (10800/3600 = 3 – szarża trwa

Przykład 4 W reaktorze rurowym, na katalizatorze, prowadzi się reakcję: k 2 CH 3 CH 2 OH (CH 3 CH 2)2 O + H 2 O k_ (VII) Szybkość rozkładu alkoholu (A) jest opisana równaniem kinetycznym: (1) B - eter, C – woda K, KA, KB i KC : stała równowagi reakcji oraz stałe równowagi adsorpcji A i desorpcji B i C; k - stała szybkości reakcji z lewej strony na prawą. Wartości stałych w 290 K i pod ciśnieniem 0, 2 MPa: k = 1, 2 10 -10 mol g-1 Pa-1 s-1; KA = 2, 9 10 -5 Pa-1; KB = 8, 7 10 -8 Pa-1; KC = 6, 3 10 -5 Pa-1; Obliczyć ilość katalizatora niezbędną do przemiany 70% substratu A podawanego ze strumieniem masy równym 23 g-1. Założyć model reaktora tłokowego.

Rozwiązanie Z równania ciągłości składnika A w stanie ustalonym w reaktorze tłokowym otrzymujemy: d. FA+rd. W = 0 FA = FA, 0(1 - ) - strumień molowy substratu A, W - masa katalizatora (2) (3) (porównać z równaniem określającym objętość reaktora tłokowego: . podstawowe równanie reaktora z przepływem tłokowym. Określa związek pomiędzy stopniem przereagowania i umownym czasem przebywania reagentów w reaktorze). W warunkach zadania strumień zasilania i stopień przereagowania wynoszą: FA, 0 = 23/46 = 0, 5 mol s-1 (Mcz. Et. OH) ; pi = pxi (xi - ułamek molowy składnika i) (ponieważ x. A, 0 = 1. (4)

Na podstawie tych zależności szybkość reakcji można wyrazić jako funkcję stopnia przereagowania. Masę katalizatora obliczamy wówczas z zależności: (5) Po podstawieniu wartości liczbowych i ciśnienia 0, 2 MPa oraz obliczeniu (numerycznym) całki otrzymuje się: Wymagana masa katalizatora wynosi 853 kg.

Przykład 5 Reakcja chlorhydryny z wodorowęglanem sodu: k CH 2 OH • CH 2 Cl + Na. HCO 3 CH 2 OH • CH 2 OH + CO 2 + Na. Cl w roztworze wodnym jest nieodwracalną reakcją o równaniu kinetycznym: A = CH 2 OH-CH 2 Cl; B = Na. HCO 3; C = CH 2 OH-CH 2 OH Stała szybkości k w 350 K wynosi 1, 44 10 -6 m 3 mol-1 s-1 Produkt C ma być wytwarzany w ilości FC = 0, 5 mol-1 s-1 (111, 6 kg h-1) z substratów podawanych w proporcji stechiometrycznej. Stężenia substratów w pierwotnych roztworach wodnych wynoszą: 30% mas. A i 15% mas. B. Założyć stałą gęstość roztworu reagującego = 1, 02 g cm 3 w 350 K.
 zbiornikowym z doskonałym mieszaniem,")
Obliczyć objętość roboczą reaktora, jeżeli proces prowadzi się reaktorze: A) zbiornikowym z doskonałym mieszaniem, B) przepływowym z doskonałym mieszaniem, C) przepływowym tłokowym, zakładając, że stopień przereagowania obu substratów wynosi 95%.
 moli A.")
Rozwiązanie Stężenie początkowe substratów: 1 g roztworu chlorhydryny zawiera 30/(80, 5 100) moli A. 1 g roztworu wodorowęglanu zawiera 157(84 100) moli B. Oba roztwory należy zmieszać w takiej proporcji, aby liczba moli A i B była równa: n. A, 0 = n. B, 0 Tak więc należy zmieszać x gramów roztworu chlorhydryny i y gramów roztworu węglanu w proporcji: aby otrzymać 1 kg roztworu, czyli: x + y = 1000 Skąd: x = 323, 944 g, w tym 1, 207 mola A; y = 676, 056 g, w tym 1, 207 mola B; w objętości 1000/1020 = 0, 9804 dm-3. Stężenie początkowe obu substratów wynosi: Roztwory pierwotne substratów należy mieszać (na doprowadzeniu do reaktora) w proporcji masowej x(A)/y(B).

Reaktor o pracy okresowej Przy zadanym stopniu przereagowania substratu czas t potrzebny do uzyskania takiej przemiany określa równanie (podające czas reakcji potrzebny do osiągnięcia stopnia przereagowania k) - niezależnie od objętości roboczej aparatu: (6) Oprócz czasu przebiegu reakcji t potrzebny jest pewien czas tj na napełnienie reaktora i na jego opróżnienie. Przyjęto, że okres ten nie zależy od wielkości reaktora i wynosi 1800 s. Aby uzyskać żądaną produkcję Fć, musi być spełnione warunki: • liczba moli produktu, otrzymana z reaktora o objętości V w czasie (t + tj) jest równa Vc. A, 0 , • w jednostce czasu należy wyprodukować FC moli produktu, zatem: Vc. A, 0 - t + tj FC 1 (7) a niezbędna objętość aparatu wynosi:

W warunkach zadania otrzymuje się kolejno: oraz Objętość reaktora zbiornikowego o pracy okresowej wynosi 5, 35 m 3.

Reaktor zbiornikowy przepływowy z doskonałym mieszaniem W warunkach pracy ustalonej objętość reaktora jest wprost proporcjonalna do strumienia objętości, stężenia początkowego substratów i stopnia przereagowania oraz odwrotnie proporcjonalna do szybkości reakcji: gdzie szybkość reakcji odpowiada końcowemu stopniowi przereagowania k Strumień objętościowy q wyznaczamy z warunku zadanej produkcji: (8) Otrzymujemy: Objętość reaktora zbiornikowego przepływowego z doskonałym mieszaniem musi być kilkanaście razy większa od objętości reaktora zbiornikowego okresowego z doskonałym mieszaniem !

Reaktor przepływowy tłokowy W warunkach stanu ustalonego objętość reaktora tłokowego oblicza się z odpowiedniego równania: Spośród trzech rozważanych reaktorów doskonałych najmniejszą objętością charakteryzuje się reaktor typu tłokowego.
 roztworem wodorotlenku sodu (B): CH 3 COOCH(CH")
Przykład 6 Kinetykę zmydlania octanu izopropylu (A) roztworem wodorotlenku sodu (B): CH 3 COOCH(CH 3)2 + Na. OH (CH 3)2 CHOH + CH 3 COONa opisano równaniem: (9) Wartość stałej szybkości k = 2, 55 10 -5 m 3 mol -s-1. Obliczyć stężenie octanu i stopień jego rozkładu na wylocie z reaktora przepływowego o następującej funkcji gęstości rozkładu czasu przebywania elementów płynu w aparacie: t, s 440 480 520 560 600 640 E(t) 104, s 0 2, 61 6, 53 18, 94 21, 23 23, 51 t, s 680 720 760 800 840 880 E(t) 104, s t, s 28, 09 30, 05 26, 78 22, 86 19, 92 920 960 1000 1040 1080 Reaktor jest zasilany roztworem o stężeniu: c. A 0 = 200 mol m-3, c. B 0 = 200 mol m-3 E(t) 104, s 9, 80 5, 42 1, 31 0, 33 0
 wykorzystującą bezpośrednio funkcję gęstości")
Rozwiązanie Dla rozwiązania założono model całkowitej segregacji z zależnością (10) wykorzystującą bezpośrednio funkcję gęstości rozkładu czasu przebywania. (10) Zależność stężenia substratu od czasu We (10) występuje wyrażenie określające stężenie substratu c. A(t) w zależności od czasu, w warunkach doskonałego mieszania w elemencie płynu. Aby otrzymać to wyrażenie należy scałkować równanie kinetyczne. W tym celu należy najpierw wyrazić stężenie substratu B przez stężenie substratu A: gdzie Z równania kinetycznego reakcji drugiego rzędu otrzymujemy: (11)
: Wyrażenie to można przekształcić do postaci wymaganej we wzorze (10):")
po obustronnym scałkowaniu (11): Wyrażenie to można przekształcić do postaci wymaganej we wzorze (10):
")
Obliczenie średniego stężenia substratu Średnie stężenie substratu A w warunkach dynamiki określonej funkcją E(t) wyraża wzór (10): Całkę tę można obliczyć w sposób przybliżony jako sumę: ( t = 40) Po podstawieniu otrzymujemy c. A, śr = 12, 25 mol m-3, czyli stopień przereagowania wynosi: Stężenie substratu B jest wtedy równe 112, 25 mol m-3.

Gdy założyć przepływ tłokowy ze średnim czasem przebywania ocenionym na podstawie funkcji gęstości czasu przebywania: wynoszącym 723 s, to oszacowane stężenie substratu A wynosi 11, 8 mol m-3. Odpowiada to stopniowi przereagowania = 94, 1%

Przykład 7 Mieszanina cząstek stałego reagenta o wymiarach: 30% cząstek o promieniu 50 m, 40% - 100 m, 30% - 200 m przepływa przez reaktor ruchem tłokowym, oddziałując z gazem. Założono, że: - wielkość cząstki nie ulega zmianie, - skład gazu jest taki sam w całej objętości, - szybkość przemiany jest limitowana przez zachodzącą reakcję chemiczną. Czas całkowitego przereagowania cząstki tz wynosi odpowiednio 300, 600 i 1200 s. Określić stopień przereagowania reagenta stałego po czasie 480 s.

Rozwiązanie W reakcji gazowego reagenta A z ciałem stałym B zgodnie z równaniem stechiometrycznym: A + BB produkty założono, że reakcja jest zapoczątkowywana na zewnętrznej powierzchni ciała stałego. Strefa reakcji stopniowo przemieszcza się w głąb ziarna. Ponadto założono, że kuliste cząstki o promieniu R nie zmieniają swojego kształtu. Najpierw wyprowadzimy równanie określające szybkość procesu, następnie wyznaczymy stopień przereagowania cząstek o określonym wymiarze a na końcu uśredniony stopień przereagowania.

Szybkość procesu Zakłada się, że szybkość dyfuzji składnika gazowego poprzez warstwę przyścienną gazu oraz szybkość dyfuzji tego składnika poprzez warstwę przereagowanego substratu stałego jest duża: o szybkości przemiany decyduje reakcja chemiczna. Stężenie substratu A na powierzchni cząstki c. A, s jest równe stężeniu A w fazie gazowej c. A, g, a na powierzchni nieprzereagowanego jeszcze jądra (o promieniu rc) stężenie substratu gazowego jest równe zeru. Szybkość procesu odniesiona do powierzchni nieprzereagowanego jądra wynosi: ks - stała szybkości reakcji powierzchniowej, m s-1, c. A, g = c. A, s - stężenie reagenta A w strumieniu gazu i na powierzchni cząstki, mol m-3

Zmniejszenie się promienia rc o wartość drc jest równoznaczne z przemianą d. NB moli stałego substratu: Podstawiając to wyrażenie do równania określającego szybkość procesu otrzymujemy: (1) B - stężenie molowe substratu B, mol/m 3 W równaniu (1) szybkość procesu jest określona przez szybkość zmiany promienia rc.
 Jest to stosunek")
Stopień przereagowania substratu stałego Stopień przereagowania określono jako: (7. 3. 25) Jest to stosunek tej części objętości cząstki, w której przemiana już zaszła do całko witej objętości cząstki. Średni stopień przereagowania Niech F oznacza masową szybkość zasilania reaktora fazą stałą. F(Ri) jest tą jej częścią, która jest złożona z cząstek o wymiarze rj: (7. 3. 26) Każda cząstka - niezależnie od jej rozmiaru - przebywa w reaktorze przez taki sam czas. Stopień przereagowania a. K(R) jest związany z czasem / zależnością otrzy maną po scałkowaniu równ. (7. 3. 24): (7. 3. 27) Czas potrzebny na całkowitą przemianę cząstki (rc = 0), wynosi: /, = P» R (7. 3. 28) Stosunek czasów t i t: jest związany ze stopniem przereagowania zależnością:

gdzie l - tf# odpowiada stosunkowi objętości nieprzereagowanego jądra do objętości cząstki. Przy różnym wymiarze cząstek stałego reagenta średnią wartość stopnia przere agowania można oszacować z równości: f(r ) l - «/, > = Z (l - a. B(Rj))-±^ (7. 3. 30) F j przy czym sumowanie obejmuje cząstki większe od tych, które ulegną pełnemu przereagowaniu. W warunkach zadania: przy czym (7'3'31) Po podstawieniu wartości liczbowych jako wynik końcowy otrzymuje się: ob sv = i _ o/l - 4 M Y _ Q 3 f j _ _48 Q_V = o, 932 v 600; l 1200; Średni stopień przereagowania wynosi 93, 2%. Przykład 7. 8. W procesie prażenia siarczku żelaza czas całkowitego przereagowania cząstki jest proporcjonalny do promienia. Założono, że obróbce będą poddane cząstki jed nakowej wielkości. Czas pełnego przereagowania /, = 1200 s. Określić ułamek nieprzere agowanego siarczku w produktach opuszczających reaktor po średnim czasie przebywania 3600 s. Reakcja zachodzi w warunkach doskonałego mieszania. Rozwiązanie. Rozważane warunki odpowiadają warunkom w uproszczonej war stwie fluidalnej. Średni ułamek nieprzereagowanego reagenta stałego B dla modelu całkowitej segregacji można określić z równania (7. 2. 19):

cesu jest reakcja chemiczna, to czas t: jest proporcjonalny do wielkości promienia. Przyjęto więc zależność (7. 3. 29): Po scałkowaniu równania określającego średnią wielkość przemiany otrzymuje się: , 2 l - exp (7. 3. 33) Dla wartości r/tz = 3600/1200 = 3, uzyskuje się l - a^&r = 0, 105. Gdyby stadium limitującym była dyfuzja gazu przez warstwę przereagowanego ziarna, wówczas t: byłby proporcjonalny do kwadratu promienia ziarna. Inna i znacznie bardziej skomplikowana będzie funkcja

Przykład 7. 9. Badano charakterystykę dynamiczną reaktora przepływowego prze znaczonego do produkcji glikolu etylenowego. W trakcie pomiaru reaktor zasilano czystą wodą w ilości takiej jak przewidywany sumaryczny strumień reagentów. W odpowiedzi na zakłócenie impulsowe w chwili t = O uzyskano następujący rozkład stężenia niereagującej substancji wzorcowej w strumieniu wylotowym C(f): Tabela 7. 1. Dane do przykładu 7. 9 /, s C(t), mmol-m"3 £(0 -1 03, s~' 0 4 3, 61 4 10 9, 01 8 29 26, 14 12 36 32, 45 16 46

koniec

Właściwości substancji chemicznych

Równowaga chemiczna

Przykład 8 Piroliza benzenu zachodzi z tworzeniem di i trifenylu zgodnie z reakcjami: k 1 2 C 6 H 6 C 12 CH 10 + H 2 k 1 k 2 C 6 H 6 + C 12 CH 10 C 18 CH 14 + H 2 k 2 Na podstawie wyników pomiarów stężeń benzenu x 1 i difenylu x 2 (x ułamek molowy), w reaktorze typu tłokowego, określić wartości stałych szybkości k 1 i k 2. Stałe równowagi wynoszą K 1 = 0, 242, K 2 = 0, 428. xi – ułamki molowe: 1 C 6 H 6; 2 – C 12 H 10; 3 – C 18 H 14 Model kinetyczny procesu:


2. Potrzebne dane określające zależność szybkości reakcji od stęże aia można otrzymać metodą graficzną. Nanosi się na wykres doświadczal nie wyznaczone wartości (c. q o ) względem czasu, a następnie ustala nachylenie stycznej do wykresu w każdym punkcie. Ocenę szybkości można też uzyskać algebraicznie przez obliczenie stosunku Wykonując takie obliczenia dla danych zestawionych w tab. 3*2 otrzymano następujące wyniki (tab. 3. 4) • Na podstawie tych danych można stwier dzić, że tylko w reakcji drugiego rzędu otrzymana wartość k nie wyka zuje dużych zmian. Brednia wartość kg, z wyników zestawionych w tabe li 3. 4, wynosi 2, 14. 3* Czas połowiofcsnej przemiany reakcji n 1 ego rzędu jest funkcją k i początkowego, stężenia caq 1 n" ( v. A)k 0 '1/2 * r^r W reakcji pierwszego rzędu In 2 ; A (równanie kinetycznej r W przykładzie wykorzystano czasy połowicznej przemiany otrzymane kolejno w jednym doświadczeniu, jak dla wyników przedstawionych w tabe li 3. 2. 1500 s , 3000 s , 3 '1/2 51/2 gdy «AO » 0, 304 kmol» m" , >. 'n « 0, 152 kmol*nr 3 ,
- Slides: 119