Dficits immunitaires de lenfant DCEM 2010 Rfrence dficits

















 - Hospitalisé à 2")


 : âge moyen")

 Allohémagglutinines")

 -précoces -Germes intracellulaires -Virus ++")






: Etude CD 40 L et expression des molécules HLA normale")



")



 • Incidence: 1/ 200 000 • Infections graves et récidivantes.")

: Pneumopathie : Aspergillus (41%) Abcès : sous-cutané : Staphylococcus spp (27%),")



 • • – Hb <9")





")
 • Phénotypage lymphocytaire : 4300")

 • Conditionnement myéloablatif : Busulfan, Endoxan, SAL • Infection respiratoire motivant un")


- Slides: 58
Déficits immunitaires de l’enfant DCEM 2010 Référence déficits immunitaires : optimiser leur approche Revue du praticien 15 octobre 2007
DEFICITS IMMUNITAIRES • Plus de 130 déficits connus, mais maladies rares • Fréquence : 1/5000 naissances • Provoquent infections, auto-immunité, allergie et cancers • Découverte des gènes impliqués : meilleure compréhension du Système Immunitaire
Immunité • Immunité innée : - Cellules NK - Cellules phagocytaires (avec récepteurs toll-like (TLR) qui reconnaissent des motifs microbiens) - Complément - Cytokines pro-inflammatoires • Immunité acquise: - facteurs cellulaires : lymphocytes T: Ag présenté en association avec CMH - Facteurs humoraux : lymphocytes B produisant les Ac
Origine des cellules du système immunitaire
Fonctions des lymphocytes T
Développement des lymphocytes B
Quand évoquer le diagnostic de déficit immunitaire ? • Infections = mode de révélation habituel, mais expression clinique très variable • Répétition et précocité des infections Infection à germe opportuniste (CMV, pneumocystis. . ) • Syndrome d’activation macrophagique, déficits sydromiques • Schématiquement : - Infections bactériennes à germes encapsulés : pneumocoque, haemophilus, pseudomonas dans les déficits humoraux et déficits en protéines du complément - Infections parasitaires (pneumocystis), fongiques et virales ou à mycobactéries dans les déficits de l’immunité cellulaire
L'interrogatoire de la famille et du carnet de santé • généalogie familiale • cancers, • histoire familiale de décès dans la petite enfance • parents apparentés Vaccinations: réactions éventuelles au BCG (vaccin vivant. . . )
Comment explorer un déficit immunitaire ? • Examens simples orientant le diagnostic - Numération formule sanguine : chiffre de lymphocytes, de polynucléaires, de plaquettes. Attention à l’hyperlymphocytose physiologique du jeune enfant ! - Dosage de Immunoglobulines : âge de l’enfant ! - Phénotypage des lymphocytes circulants
Comment explorer un déficit immunitaire ? • Examens plus spécifiques : - Sous classes d’Ig - Etude fonctionnelle des lymphocytes T: tests de prolifération avec mitogènes - Etude fonctionnelle des polynucléaires - Myélogramme (ex : neutropénie) - Etude du complément
Radiographie du thorax (voit on l’ombre Rx Thoracique du thymus: pneumopathie interstitielle ? Ombre thymique? existe-t-il une pneumopathie interstitielle ? …).
Quand suspecter une anomalie des LB ?
Maladie de Bruton • Cas cliniques: Enzo, né en 11/04 Pas d’antécédents familiaux (2 frères de la mère en bonne santé) Peu de problèmes infectieux : - 11/06 : anite + angine streptococcique - 01/08 : otite G - 03/08 : pneumopathie lobaire Dte
Diagnostic et Tt • Dosage des Immunoglobulines montrant Ig. G < 0, 5 g/l • Profil lymphocytaire : pas de lymphocytes B, mais lymphocytes T normaux • Explorations complémentaires : mutation de Btk chez l’enfant et sa mère • Début de Tt immédiat par Ig IV puis Ig sous cutanées
Brice né le 07 août 98 Hospitalisation à 23 mois pour fièvre et neutropénie Interrogatoire - 2 iéme enfant d’un couple non consanguin d’origine française - 1 sœur aînée de 8 ans qui va bien - 1 oncle maternel décédé à l ’age de 11 ans d’infection sévère
Antécédents personnels - Vaccins à jour - Hospitalisé à 3 reprises : à 16 mois : méningite présumée virale à 19 mois : fièvre à 40°C et grand Sd inflammatoire (CRP = 220 mg/l) non documentée à 20 mois : nouvelle méningite présumée virale - Nombreux épisodes d ’infections rhinopharyngées justifiant d ’une antibiothérapie.
Ludovic né le 28 mars 92 Hospitalisation à l ’age de 4 ans 1/2 pour pneumopathie récidivante et suspicion de déficit immunitaire Interrogatoire - 2 iéme enfant d’un couple non consanguin d’origine française - 1 sœur aînée de 9 ans qui va bien - 1 oncle maternel décédé à 3 ans d ’une méningite
Antécédents personnels - Vaccins à jour (Tétracoq, ROR, BCG) - Hospitalisé à 2 reprises : à 12 mois : diarrhée aiguë à Rotavirus à 38 mois : pneumopathie non documentée = Sd inflammatoire : CRP = 260 mg/l = Leuconeutropénie : GB : 3800/mm 3(1% de PNN) - Nombreux épisodes d ’infections rhinopharyngées justifiant d ’une antibiothérapie
Cliniquement - Courbe staturo-pondérale normale - Fièvre et symptomatologie pulmonaire avec ronchus et râles bilatéraux - Diarrhée aiguë fébrile Biologie - Grand Sd inflammatoire - Neutropénie isolée : 360 /mm 3 (Ludovic) - 38/mm 3 (Brice) Bactério - Pseudomonas aeruginosa (selles + gorge) Brice - pas de documentation bactério (sous ATB)
Bilan immunologique : Brice Ig. A < 0, 06 Ig. G < 0, 33 Ig. M 0, 05 totaux (/mm 3) 4070 B CD 19+ (%) <1 Test de stimulation n° Ac vaccinaux absents Ludovic <0, 06 0, 09 1, 26 6307 <1 n° absents
Agammaglobulinémie liée à l’X • Otites chroniques, infections bactériennes récidivantes (pulmonaires) : âge moyen de diagnostic 30 à 40 mois (pour 10% diagnostic après 5 ans) • Parfois + précoce, cellulite, abcès, septicémie, neutropénie (défaut de maturation des PNN en situation de stress) • Pas de gg cervicaux • Déficit de développement des cellules B, mais chez quelques patients, Ig G , Ig M ou Ig. A résiduelles • Réponse vaccinale nulle.
Agammaglobulinémie liée à l’X • Diminution des cellules B circulantes : cellules CD 19 + <0, 1% • Moelle osseuse : présence de cellules précurseurs B, mais pas de cellules pré-B , ni de cellules B matures • Infections : H. influenzae, S. pneumoniae, mycoplasmes, giardiase, infections chroniques à enterovirus • Meilleur pronostic actuel avec Ig IV/SC
Immunité Humorale : Bilan • • • Dosage pondéral des Ig sériques (normes) Allohémagglutinines de groupe Ac vaccinaux Sous-classes Ig Déficit cellulaire associé
Déficits Humoraux : traitement • TT Substitutif par perfusions d ’Ig/Ig sous cutanées, avec maintien de taux élevé 68 g/l • Antibiothérapie précoce et prolongée • Antibiothérapie alternée au long cours • Kinésithérapie respiratoire • Bactrim si déficit T associé • Drainage chirurgical d ’un foyer infectieux (sinus)
Quand suspecter une anomalie des LT (immunité adaptative) -précoces -Germes intracellulaires -Virus ++
Déficits immunitaires T • Anomalies de nombre ou de fonction des Cellules T • Infections répétées avec retard staturopondéral • Autoimmunité : hémato, colites, hépatites • Déficit HLA II, Déficit cytokines, Déficit CD 3, Déficit ligand CD 40 (hyper Ig. M). . .
Déficit T : traitement • Traitement préventif et curatif des infections bactériennes, virales et fongiques • Nutrition • Transplantation de cellules souches hématopïétiques avant complications
Cas clinique : Hugo né le 25/09/96 • Pneumonie février 1998 • Pneumopathie traînante mars 1998 • Epiglottite avec 1 hémoculture positive à Haemophilus juillet 1998 • Bilan immunitaire Ig G nles, Ig. M élevées, diminution des lymphocytes T • Ig. G: 6. 44 g/l, Ig. A: 0. 90 g/l, Ig. M: 3. 33 g/l déficit en Ig. G 2 et Ig. G 4 • Vaccinations non faites (1 seule injection DT polio)
Evolution • Vaccins non faits malgré prescription pour étude réponse vaccinale • Persistance d’infections ORL et pulmonaires pendant l’hiver : Tt ATB, kinésithérapie • Scanner thoracique en 2000 : atélectasies du lobe moyen et du LIG, pas de DDB • Décision de débuter un Tt par Ig IV, car hyper Ig. M et déficit en sous classes Ig. G 2 et Ig. G 4 • Début du Tt par Bactrim car aggravation de la lymphopénie T
Bilan immunologique • Hyper Ig. M, avec déficit en sous classes d’Ig. G, absence de réponse Ac à Haemophilus et pneumocoques malgré infections répétées • Altération des fonctions T correspond habituellement à un déficit en CD 40 ligand • Pas d’anomalie moléculaire décelée chez Hugo • Mucovisidose et dyskinésie ciliaire éliminées
Différents bilans: 08/98 03/99 04/00 09/00 10/02 07/05 12/05 LT 2730 2520 1710 1860 1937 3547 1128 LT 4 850 833 530 391 329 355 250 Ig. G Ig. M 5. 31 2. 83 - 7. 97 4. 74 - - 11. 1 1. 25 - -
Bilans étiologiques (C. Malcus): Etude CD 40 L et expression des molécules HLA normale en 2000 Pas de déficit en ADA ni en PNP Présence de CD 4/CD 45 RA+ et CD 31 + à 6% témoins d’une thymopoïèse insuffisante Absence d’anomalie de répartition des familles Vβ sur CD 4 et CD 8 Répertoire T normal
Evolution ultérieure: Poursuite des Ig IV et du Bactrim Survenue d’une dilatation des bronches : suivi en pneumo avec kiné respiratoire, antibiothérapie alternée -Juin 04: apparition d’une diarrhée: cryptosporidiose. -Février 05: polyadénopathie périphérique et abdominale: biopsie ADP: ADP réactionnelle Hypotrophie sévère -Novembre 05: hospitalisé en réanimation pour deshydratation aigue et intoxication à l’eau (Hypo. Na 117, Hypo. K 2. 3, perte de 14% du poids du corps) sur récidive de la cryptosporidiose. Dénutrition. Echographie abdominale: pneumatose du grèle, aspect de cholangite sclérosante et persistance des ADP abdominales.
Fin 2005: Cliniquement: dénutri, HMG à 6 cm, ballonnement abdo, en AP complète pour cryptosporidiose sévère Biopsie hépatique: fibrose portale, pas de cirrhose, pas de cholangite sclérosante. PCR EBV en augmentation: 16 750 copies Colonisation digestive à ADV de type 2 PCR CMV positive dans le sang mais non quantifiable (< 500) Allogreffe ? Conditionnement atténué ?
Greffe de cellules souches hématopoiétiques • Cordon 4/6ème identique, riche en l’absence de donneur familial • Conditionnement « réduit » : Fludarabine, SAL, Melphalan • Prévention infectieuse « musclée » : flux laminaire, Bactrim, Ig. G IV hebdomadaires, Cidofovir (adénovirus et cytomégalovirus, anti. CD 20 pour éviter réactivation de l’EBV et alinia+azithromycine (cryptosporidiose)
Evolution post greffe • Reconstitution hématologique à J 30 avec chimérisme complet (100% donneur) • Seule complication : réactivation du CMV traitée par antiviraux • Arrêt de la majorité des Tt préventifs à 6 mois post greffe du fait de la bonne reconstitution immunitaire; Reprise des vaccinations • Très bon état général à 3 ans post greffe. Scolarisation normale
Déficits des PN ? Ex Neutropénies
Neutropénies constitutionnelles • • Maladies hétérogènes, rares Blocage de la myélopoïèse Mutations de gènes ELA 2, HAX 1, WAS, … Amélioration du pronostic depuis l’utilisation des facteurs de croissance (GCSF) • Beaucoup plus rares que les neutropénies auto-immunes de la petite enfance!
Déficits des PN/ macrophages ? Déficits de la bactéricidie
Granulomatose septique chronique (CGD) • Incidence: 1/ 200 000 • Infections graves et récidivantes. • Anomalies du système de la NADPH oxydase (phagocytose). • Amélioration considérable du pronostic: Prise en charge plus précoce, plus agressive prophylaxie au long cours
Manifestations cliniques: • Fonction de la localisation des granulomes. • Infections récidivantes ++ • Apparition précoce: 70% avant 1 an. Pneumopathies 79% Abcès 68% Adénites suppuratives 53% Ostéomyélites/ arthrites 25% Septicémies 18% Otites 15% Cellulites 5% Méningites 4% Divers: sténoses T. D, HPSMG, malabsorption, dénutrition…
Manifestations cliniques (suite): Pneumopathie : Aspergillus (41%) Abcès : sous-cutané : Staphylococcus spp (27%), hépatique : Staphylococcus spp (50%) poumon : Aspergillus (23%) cérébraux : Aspergillus (58%) Adénite suppurative : Staphylococcus spp (26%) Ostéomyélite : Serratia (29%), Aspergillus (22%) Septicémie : Salmonelle (18%), Burkholderia (12%), Candida (11%) Méningite : Candida (20%) Aspergillus est responsable 1/3 des DC
Traitement • • Préventif : Bactrim, antifongiques Curatif : anti infectieux Greffe de cellules souches hématopoïétiques Thérapie génique ?
Syndromes d’activation macrophagique • Activation et prolifération non maligne des Lymphocytes T et des Macrophages. • S’accompagnant d’une Hypercytokinémie rendant compte des principaux signes Cliniques et Biologiques
SAM: Signes Cliniques • • Fièvre Splénomégalie HPG, ADP. . Altération E. G. Eruption cutanée (rash) oedèmes F. graves: Syndr. hémorragique, troubles de la conscience/ convulsions… • Autres dysfonctionnements d’organes
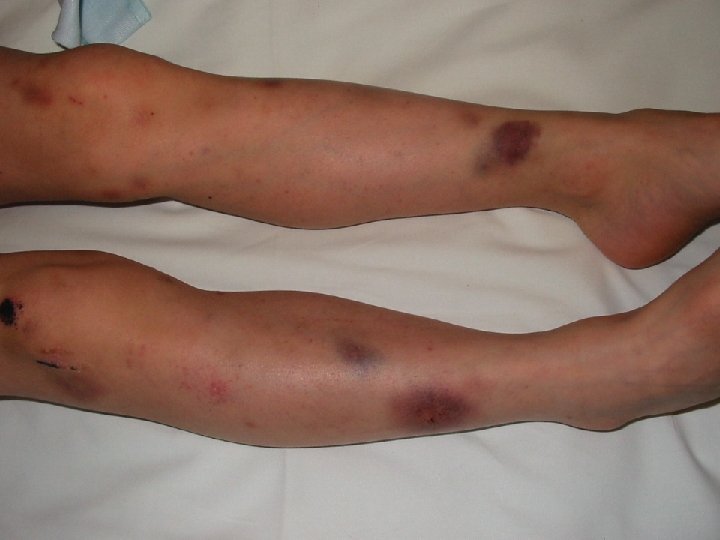
SAM: Signes Biologiques • Cytopénie (>2 ou 3 lignées) • • – Hb <9 g/dl. – Neutropénie <1000/mm 3 – Thrombopénie <100. 000/mm 3 Hypertriglycéridémie (>3 DS), Fibrinopénie (<3 DS) Cytolyse hépatique Baisse II, VII, X, (V). C. I. V. D. Augmentation LDH, Ferritine. Hyponatrémie Lymphocytes T activés
SAM: Signes Histologiques • Atteinte Moelle osseuse: Hémophagocytose • Infiltration des organes lymphoïdes par Lymphocytes et Macrophages. Pas de signe de malignité
Syndrome d’activation macrophagique: Prédispositions constitutionnelles et acquises Acquises Constitutionnelles Anomalies immunitaires constitutionnelles - Lymphohistiocytoses F. - Maladie de Chediak-Higashi - Maladie de Griscelli - Maladie de Purtilo Médicaments Hémopathies M Lymphome T EBV+ Histiocytose L. Infections Maladies métaboliques Intolérance aux protéines dibasiques Maladies auto-immunes M. inflammatoires ou rhumatismales
Conclusion: SAM • Syndromes hémophagocytaires: rôle déterminant de la fonction cytotoxique des Lymphocytes T. • Diagnostic moléculaire/ Conseil génétique • Traitement: Immunosuppresseur ciblant les lymphocytes T/ greffes pour les formes génétiques
Déficits immunitaires ‘syndromiques’ Ataxie télangiectasie
Syndrome de Wiscott Aldrich • Cas clinique : Enzo, né en juillet 2001 • Premier enfant d’un couple non consanguin • Eczéma à 1 mois traité par corticoïdes locaux • Thrombopénie à 3 mois de vie, associée à une altération de l’état général
Cas clinique • • - Examen : taille et PC Nx, poids (-1 DS) Polypnée, tirage intercostal (Sat 92%) Pétéchies, quelques lésions d’eczéma Examens : Thrombopénie 37 G/L avec plaquettes petites (5 fl) - Rx thorax : syndrome interstitiel - LBA: pneumocystis, staphylocoque doré, Klebsielles, candida parapsilosis
Bilan immunitaire • Hypergammaglobulinémie (Ig G et Ig M) • Phénotypage lymphocytaire : 4300 Ly avec 53% CD 3, 48% CD 4, 4% CD 8 • Proliférations satisfaisantes avec PHA, nulles avec tétanos (non vacciné) • Coombs positif
Traitement • Antibiothérapie adaptée et traitement antifongique • Isolement en chambre stérile • Greffe de cellules souches hématopoïétiques à partir d’un donneur non apparenté après amélioration de l’état pulmonaire
Traitement (suite) • Conditionnement myéloablatif : Busulfan, Endoxan, SAL • Infection respiratoire motivant un séjour en réanimation pendant 1 semaine à partir de J 7 • Gv. H hépatique imposant l’accentuation de l’immunosuppression • Chimérisme partiel, avec persistance de la thrombopénie , mais bon développement immunitaire
Del 22 q 11 : syndrome de Di George
Conclusions • Nombreux gènes connus DG Précoce • Lutte rapide et efficace contre infections • Transplantation de moelle osseuse dans DICS et déficits T , le + tôt • Thérapie génique ? • Dg anténatal