Potencjometria Metody analityczne wykorzystuj cztery grupy reakcji chemicznych

 2. reakcje kompleksowania(kompleksometria) 3. reakcje strącania osadów (precypitometria) 4.")


")




 Przed osiągnięciem punktu równoważności potencjał roztworu zależy tylko od potencjału układu miareczkowanego: Utl")
 Potencjał redoks układu po przekroczeniu punktu równoważności Potencjał w dowolnym punkcie krzywej miareczkowania")

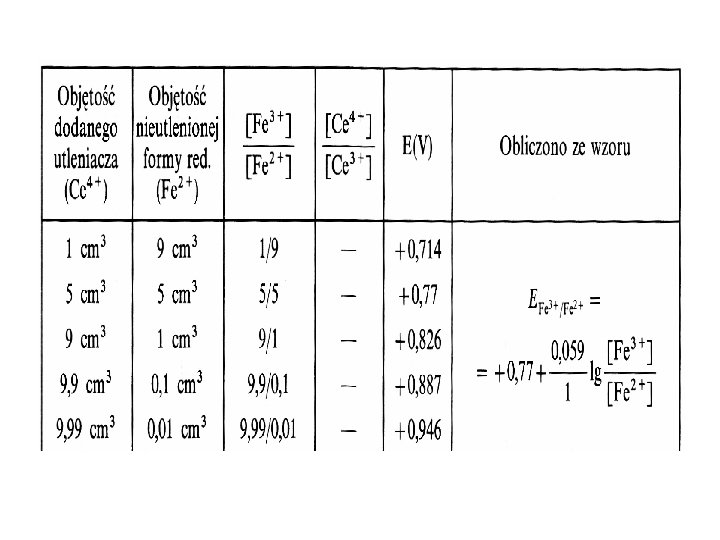

 Potencjał układu w punkcie równoważności W punkcie równoważności (PR) potencjały obu układów")
 •")
 - rzeczywisty potencjał normalny")



’+")









 podczas miareczkowania")







![[Ag. Cl]=[Ag+]=[Cl-]= 0, 01 mol/l W temperaturze 25 o. C obliczony potencjał elektrody chlorosrebrowej](https://slidetodoc.com/presentation_image_h2/8adfdd64b80eb568069a79e689c00bc9/image-40.jpg "[Ag. Cl]=[Ag+]=[Cl-]= 0, 01 mol/l W temperaturze 25 o. C obliczony potencjał elektrody chlorosrebrowej")

 roztwór Na. I+stały")
![Aby obliczyć stężenie [I-], należy uwzględnić fakt, że Na. I Na+ + I-: n](https://slidetodoc.com/presentation_image_h2/8adfdd64b80eb568069a79e689c00bc9/image-43.jpg "Aby obliczyć stężenie [I-], należy uwzględnić fakt, że Na. I Na+ + I-: n")
![Ponieważ Ks. Ag. I l= 2 10 -16 = [Ag+] [ I-] , więc](https://slidetodoc.com/presentation_image_h2/8adfdd64b80eb568069a79e689c00bc9/image-44.jpg "Ponieważ Ks. Ag. I l= 2 10 -16 = [Ag+] [ I-] , więc")
- Slides: 44
Potencjometria Metody analityczne wykorzystują cztery grupy reakcji chemicznych:
1. reakcje kwas –zasada (alkacymetria) 2. reakcje kompleksowania(kompleksometria) 3. reakcje strącania osadów (precypitometria) 4. reakcje redox (redoksymetria) Reakcje redox (redoksymetria) Metody oparte na reakcjach utleniania i redukcji. • W miarę przebiegu reakcji reduktor ulega utlenieniu (oddaje elektrony), natomiast utleniacz ulega redukcji (przyjmuje elektrony). • •
• • • Oksydometria - titrant jest odczynnikiem o własnościach utleniających: manganometria KMn. O 4 Mn 7+ → Mn 6+, Mn 4+, Mn 2+ cerometria Ce(SO 4)2 Ce 4+ → Ce 3+ chromianometria K 2 Cr 2 O 7, K 2 Cr. O 4 Cr 6+ → Cr 3+ bromianometria KBr. O 3 Br 5+ → Brjodometria
Reduktometria - titrant jest odczynnikiem o własnościach redukujących (ferrometria Fe. SO 4, tytanometria Ti. Cl 3, askorbinometria ) Punkt równoważnikowy (PR) wyznaczany jest: za pomocą wskaźników i potencjometrycznie. Wskaźniki te są układami redox, których forma utleniona posiada odmienne zabarwienie niż forma zredukowana.
W miareczkowaniach manganometrycznych i jodometrycznych nie ma potrzeby stosowania dodatkowych wskaźników, gdyż manganian (VII) potasu i jod same odgrywają ich rolę. Np. jako roztworu miareczkującego używa się manganianu (VII) potasu o silnie fioletowym zabarwieniu, który w zależności od środowiska reakcji (ze zmianą p. H zmienia się potencjał redoks układu) ulega różnym przemianom: • środowisko kwaśne - roztwór ulega odbarwieniu Mn. O 4¯ + 8 H+ +5 e → Mn 2+ + 4 H 2 O (E° = + 1, 53 V) • środowisko obojętne lub słabo zasadowe lub słabo kwasowe – wytrąca się brunatny osad Mn. O 2 Mn. O 4¯ + 2 H 2 O +3 e → Mn. O 2 ↓ + 4 OH¯ (E° = + 0, 58 V) • środowisko mocno zasadowe - roztwór przybiera zieloną barwę Mn. O 4¯ + e → Mn. O 42¯ (E° = + 0, 56 V)
Miareczkowanie potencjometrycznie Reakcje utleniania i redukcji, będące podstawą miareczkowania redoksymetrycznego, przebiegają znacznie wolniej od reakcji jonowych (np. alkacymetria). Wymiana elektronów pomiędzy jonami przebiega w kilku etapach, a najwolniejszy z nich decyduje o szybkości całego procesu. Ogólne równanie reakcji redoks można schematycznie zapisać • Ox 1 + n 1 e → Red 1 | n 2 • Red 2 → Ox 2 + n 2 e | n 1 • n 2 Ox 1 + n 1 Red 2 = n 2 Red 1 + n 1 Ox 2
Układ, w którym postać utleniona jest związana z postacią zredukowaną tylko wymianą elektronów, nazywany jest sprzężoną parą redoks (np. Fe 3+/Fe 2+). Podczas miareczkowania redoks, w miarę dodawania odczynnika (utleniacza lub reduktora) zmieniają się stężenia postaci utlenionej i zredukowanej miareczkowanego układu. Mamy więc do czynienia z dwoma układami redoks, którym odpowiadają potencjały obliczone ze wzoru Nernsta:
• • Układy te reagują ze sobą zgodnie z ogólnym równaniem: m 1 Utl 1 + m 2 Red 2 m 1 Red 1 + m 2 Utl 2 gdzie: m 1, m 2 – współczynniki reakcji n 1, n 2 – liczby elektronów wymienianych w odpowiednich reakcjach połówkowych redoks m 1 – jest bardzo często równe n 1, natomiast m 2 jest bardzo często równe n 2. k – 2, 303 RT/F i wynosi: 0, 060 V w t=30 o. C; 0, 059 V w 25 o. C i 0, 058 V w 20 o. C itd. R – stała gazowa, 8, 314 J K-1 mol-1 T – temperatura bezwzględna F – stała Faradaya, 96487 C mol-1
Krzywa miareczkowania redoks jest zależnością potencjału utleniającoredukującego układu redoks od objętości dodawanego titranta E’= f(Vtitr). Krzywą miareczkowania redoks można podzielić na trzy odcinki: a) odcinek krzywej do punktu równoważności b) w punkcie równoważności (PR) c) odcinek krzywej w dowolnym punkcie miareczkowania po przekroczeniu PR.
a) Przed osiągnięciem punktu równoważności potencjał roztworu zależy tylko od potencjału układu miareczkowanego: Utl 1 + n 1 Red 1. Dowolny punkt na tym odcinku krzywej można obliczyć z równania Nernsta dla układu miareczkowanego: b) Ered 1 = Eutl 2 = EPR
c) Potencjał redoks układu po przekroczeniu punktu równoważności Potencjał w dowolnym punkcie krzywej miareczkowania po przekroczeniu punktu równoważności obliczamy podobnie jak dla przypadku przed PR. Potencjał utleniającoredukujący roztworu zależy od potencjału układu, którym miareczkujemy (titranta) Red 2 Utl 2 + n 2 Dowolny punkt na tym odcinku krzywej można obliczyć z równania Nernsta dla układu titranta:
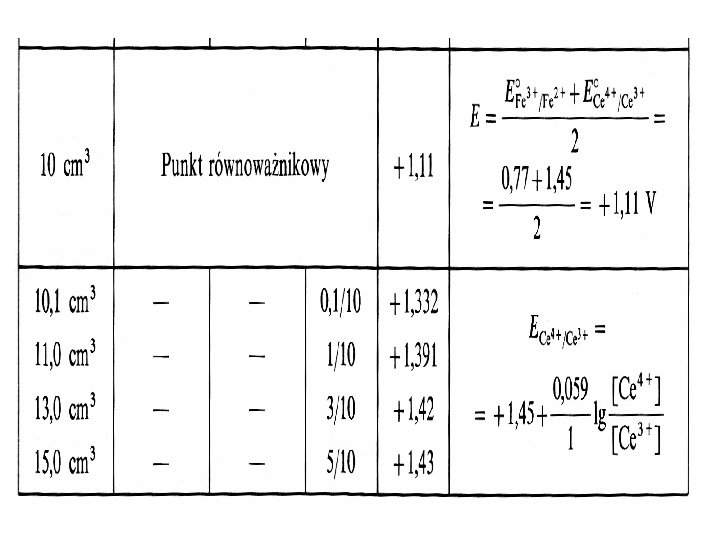
Przykładowa krzywa miareczkowania potencjometrycznego Miareczkowanie 10 ml 0, 1 mol/l roztworu jonów Fe 3+ roztworem 0, 1 mol/l jonów Ce 4+ w 1 mol/l HCl, E 0 Ce 4+/Ce 3+ = 1, 45 V, E 0 Fe 3+/Fe 2+ = 0, 77 V, temp 25 o. C (pod. red. R. Kocjana, „Chemia analityczna”, Wydawnictwo Lekarskie PZWL, W-wa 2000) W całym dziale Redoksymetrycznym ten proces nazywa się oksydometrią, a dokładniej, jeżeli titrantem jest Ce to Cerometrią Cr 4+ + Fe 2+ Ce 3+ + Fe 3+ (gdyby titrantem był Fe to Ferrometria i proces ogólnie zwany Reduktometrią).
Krzywa miareczkowania jonów Fe 2+ roztworem jonów Ce 4+
Ad. b) Potencjał układu w punkcie równoważności W punkcie równoważności (PR) potencjały obu układów redoks są sobie równe: a) E 1’ = E 2 ‘= E’PR oraz b) [Utl 1] = [Red 1 ] i [Utl 2] = [Red 2]
Mnożymy odpowiednio równania przez n 1 lub n 2 oraz uwzględniamy założenie b) • E’ 1 · n 1 = E’’ 0(1) · n 1 + | k/n 1· n 1 ·log 1 • E’ 2 · n 2 = E’’ 0(2) · n 2 + | k/n 2· n 2 ·log 1 Dodajemy równania stronami: E’ 1 · n 1 + E’ 2 · n 2 = E’’ 0(1) · n 1 + E’’ 0(2) · n 2 Uwzględniając punkt a) E 1’ = E 2 ‘= E’PR (n 1+n 2) = E’’ 0(1) · n 1 + E’’ 0(2) · n 2
Ostatecznie potencjał w PR obliczamy ze wzoru: gdzie: E’’ 0(1) - rzeczywisty potencjał normalny reakcji połówkowej (1) E’’ 0(2) - rzeczywisty potencjał normalny reakcji połówkowej (2) • n 1 – liczba elektronów przenoszona w reakcji (1) • n 2 - liczba elektronów przenoszona w reakcji (2)
Potencjał rzeczywisty obliczmy w dwojaki sposób w zależności czy w reakcji połówkowej nie biorą udział jony H+ np. Fe 2+ Fe 3+ + e to potencjał rzeczywisty E’’ 0(1) = E’ 0(2) • Inna jest sytuacja, jeżeli w reakcji biorą udział jony H+ • Zad. Jaki jest potencjał półogniwa w PR miareczkowania jonów Fe 2+ jonami Mn. O 4 -- w obecności kwasu, jeżeli p. H w PR wynosi 2, 3, temp 25 o. C, E 0 Fe 3+/Fe 2+ = +0, 771 V (reduktor), E 0 Mn. O 4 -/Mn 2+ = +1, 51 V (utleniacz)
Fe 2+ Fe 3+ + 1 ē x 5 Mn. O 4 - + 5 ē + 8 H+ Mn 2+ + 4 H 2 O x 1 _______________ 5 Fe 2+ + Mn. O 4 - + 8 H+ 5 Fe 3+ + Mn 2+ + 4 H 2 O /· 5
• W celu likwidacji n w mianowniku mnożymy każde z nich przez właściwe n oraz dodajemy oba równania stronami: 6 E’ = Eo(1)’’ + 5 Eo(2) ‘’+ W PR = oraz = Czyli [Fe 3+] = 5 [Mn 2+] oraz [Fe 2+] = 5[Mn. O 4 -] w równaniu pozostają tylko zależności Mn 2+ i Mn. O 46 E’ = Eo(1)’’ + 5 Eo(2) ‘’+
Po skróceniu tych samych odpowiednich wyrazów, równanie wygląda następująco: E’ = 1/6 · (Eo(1)’+ 5 · Eo(2)’) + · log [H+]8 Ostateczne, uporządkowane równanie ogólne + · log [H+]m lub _ · p. H Obliczenie zadania E’PR = _ 2, 3 · = 1, 21 V
Potencjał PR nie zależy ani od stężeń, ani aktywności składników układu redoks biorących udział w reakcji redoks, gdyż te wielkości nie występują w powyższym równaniu. Jeżeli w reakcji redoks uczestniczą pośrednio, tzn. , jeżeli są wiązane lub wydzielane jony H+ lub OH-, to wtedy p. H wpływa na Eo, , . Wtedy należy uwzględnić wpływ tych jonów w obliczeniach: gdzie: Eo, - potencjał normalny układu redoks k – współczynnik zależny od temperatury m – współczynnik stechiometryczny n – liczba elektronów wymienionych w reakcji p. H= – log [H 3 O+]
• Potencjał normalny to stała termodynamiczna, niezależna od składu roztworu. • Potencjał formalny (rzeczywisty) to wartość stała, określona dla ściśle sprecyzowanego składu roztworu. • Dla każdej pary reakcji połówkowych w danej temp. jest tylko jeden termodynamiczny potencjał standardowy Eo ale potencjałów formalnych (rzeczywistych) może być wiele. • SEM ogniwa pomiarowego zależy od potencjału elektrody odniesienia i w przypadku zastosowania NEK (nasyconej elektrody kalomelowej), będzie mniejsze o ok. 0, 25 V od wartości E’ obliczonych względem normalnej elektrody wodorowej
• Zad. Jaki będzie potencjał rzeczywisty dla układu Cr 2 O 72 -/Cr 3+ w PR przy p. H 3, 5 w temp. 15 o. C E 0 wynosi +1, 3 V względem a) NEW b) NEK a) elektroda platynowa wg NEW Cr 2 O + 6ē + 14 H+ 2 Cr 3+ + 7 H 2 O Ponieważ [Cr 2 O ]= [Cr 3+]2 = 50% E’ = 1, 3= +0, 835 V b) względem NEK 0, 835 -0, 25 = 0, 585 V więc
1. Oblicz potencjał elektrody miedziowej zanurzonej do roztworu zawierającego 0, 01 mol/l Cu. SO 4 i 0, 01 mol/l Cu(NO 3)2 , temp. 25 o. C: a) względem nas. elektrody wodorowej b) względem nas. elektrody kalomelowej, jeżeli potencjał normalny elektrody kalomelowej wynosi 0, 34 V Cu 2+ + 2 ē Cu 0 + 1ē a) Potencjał elektrody miedziowej =0, 34 + 0, 0295 log 0, 02=0, 290 V Potencjał normalny miedzi jest wyrażony względem normalnej elektrody wodorowej i obliczona wartość jest równa względem tej elektrody. b) Potencjał nasyconej elektrody kalomelowej względem wodorowej wynosi 0, 244 V, a więc potencjał elektrody miedziowej względem nasyconej kalomelowej wynosi: +0, 290 -(+0, 244)= +0, 046 V
2. Obliczyć SEM ogniwa złożonego z dwóch półogniw redox Fe 3+/Fe 2+ i Cr 2 O 72 - i Cr 3+, jeżeli stężenia wszystkich składników układu wynoszą każdego z osobna 0, 1 mol/l temp. 25 o. C Potencjały normalne odpowiednio 0, 77 V i 1, 33 V = + 0, 77 V = SEM= E 2 -E 1 SEM=1, 20 -0, 77= +0, 43 V +1, 20 V
3. Obliczyć SEM ogniwa chemicznego złożonego z dwóch półogniw redox , temp. 25 o. C V V Pt||Fe 3+ (1 mol/l ) Fe 2+ (0, 010 mol/l ) || Ce 4+ (0, 00010 mol/l) || Pt Ce 3+ (0, 10 mol/l) Jony Ce 4+ są utleniaczem a same się redukują aż do stanu równowagi i prąd przestaje płynąć Ce 4+ + Fe 2+ Ce 3+ + Fe 3+ =+0, 889 V
= + 1, 433 V SEM= E 2 - E 1 SEM= 1, 433 -0, 889= +0, 54 V
KRZYWA MIARECZKOWANIA STRĄCENIOWEGO Miareczkowanie strąceniowe prowadzi się w układzie zawierającym trudno rozpuszczalny osad oraz metaliczną elektrodę znajdującą się w równowadze z jonami tego samego metalu, występującymi w roztworze i osadzie: Meo (elektroda) Men+ + ne, w obecności osadu. Aktywność formy zredukowanej a. Me = 1. Równanie Nernsta przybiera wówczas postać typową dla elektrody drugiego rodzaju.
W celu przeprowadzenia tego typu miareczkowania, należy zbudować ogniwo złożone z elektrody odniesienia o stałym potencjale oraz elektrody wskaźnikowej, której potencjał zależy od stężenia oznaczanego jonu, jest to jednocześnie jeden z jonów wchodzących w skład osadu. Osad ten powstaje jako produkt reakcji pomiędzy roztworem miareczkującym i miareczkowanym. Zastosowanie miareczkowania potencjometrycznego osadowego wymaga zastosowania odpowiedniej elektrody wskaźnikowej. Rodzaj elektrody wskaźnikowej zależy od składu osadu. Metodę tę wykorzystuje się najczęściej do oznaczania jonów Ag+ oraz jonów tworzących osady z jonami Ag+, takich jak Cr-, Br-, I-, SCN- i inne. •
Potencjał układu przed punktem równoważności W przypadku oznaczania Ag+ lub Cl-(X -) podczas miareczkowania zachodzi reakcja: Ag+ + X - Ag. X W roztworze zmienia się stężenie jonów Ag+ i X-, w sposób opisany przez iloczyn rozpuszczalności Ks soli tworzącej się podczas miareczkowania: Ks. Agx = [Ag]+[X -] Jako elektrodę wskaźnikową stosuje się elektrodę srebrową, która jest elektrodą metaliczną pierwszego rodzaju. Jej potencjał zmienia się zgodnie z równaniem Nernsta: Jednocześnie jest to wzór na potencjał elektrody I rodzaju: Przed PR stężenie jonów Ag+ nie związanych w osad jest dość duże w stosunku do tych, które są określone iloczynem rozpuszczalności soli Ag. X. W miarę dodawania jonów X – zmniejsza się stężenie wolnych jonów Ag+, a więc potencjał jest coraz mniejszy.
Potencjał układu w punkcie równoważności Podczas miareczkowania Ag+ jonami X – w PR wszystkie jony Ag+ są związane w osad. Stężenie jonów Ag+ jest określone przez iloczyn rozpuszczalności soli: [Ag+] = Ponieważ: Ag. X Ag+ +Xwięc Ks. Ag. X = [Ag+] · [X-] dlatego dla soli typu 1: 1, tzn. AX, stężenie obu składników w PR są jednakowe [Ag+] = [X-] =
Przy miareczkowaniu strąceniowym PR odpowiada tym stężeniom składników związku trudno rozpuszczalnego, przy których roztwór czystej substancji jest nasycony w danej temperaturze. Dla soli typu A 2 X w PR np. Ag 2 S 2 Ag+ + S 2 X 2 x x Ks= (2 x)2 · x= 4 x 3 Stąd x= wtedy [Ag+] = 2 = potencjał wynosi E’ = Eo + k/n ·log 2 · lub E’ = Eo + k/n Ogólnie dla A 2 X [A+] = 2[X-], czyli: [X -] = , a więc [A+] = 2 =
Potencjał układu po przekroczeniu PR Jeżeli miareczkujemy Ag+ jonami X -, to po przekroczeniu PR mamy układ przypominający elektrodę drugiego rodzaju, czyli odwracalną względem anionu: Ago/Ag. X (X -) elektroda odniesienia. O potencjale elektrody srebrowej decydują jony Ag+, które są określone przez Ks Ag. X oraz efekt wspólnego jonu przez jony X-, które po PR są w dużym nadmiarze. Potencjał liczymy wówczas ze wzoru:
Potencjał maleje w miarę wzrostu stężenia jonów X -, które obniżają rozpuszczalność osadu, a tym samym stężenie Ag+. W przypadku oznaczania jonów X – za pomocą Ag+ sytuacja przed i po PR jest opisana odwrotnie.
Przykłady 1. Obliczyć potencjał elektrody chlorosrebrowej zanurzonej w roztworze 0, 01 mol/l Na. Cl. Normalny potencjał elektrody chlorosrebrowej względem normalnej elektrody wodorowej wynosi + 0, 222 V Elektroda chlorosrebrowa jest elektrodą drugiego rodzaju, jest odwracalna względem anionu. Przemiany zachodzące na elektrodzie można przedstawić równaniem: Ago + Cl- Ag. Cl + 1ē W stanie równowagi szybkości reakcji w obu kierunkach są jednakowe.
Potencjał tej elektrody jest wyrażony wzorem Nernsta: lub Elektrodą chlorosrebrową jest zazwyczaj drut srebrny pokryty elektrolitycznie Ag. Cl , zanurzony w roztworze zawierającym jony Cl-, pochodzące z chlorku lub kwasu solnego. Schemat elektrochemiczny tej elektrody: Ag/ Ag. Cl(s) /Cl-.
Elektrodę chlorosrebrową można traktować jako elektrodę reagującą na aktywność jonu Ag+, która jest uzależniona od aktywności jonu Cl- przez iloczyn rozpuszczalności Ag. Cl Ponieważ Ks. Ag. Cl=1, 6 10 -10 w 25 o. C, to potencjał elektrody chlorosrebrowej, zależy od aktywności a jonu Cl-. Mamy potencjał normalny elektrody chlorosrebrowej.
[Ag. Cl]=[Ag+]=[Cl-]= 0, 01 mol/l W temperaturze 25 o. C obliczony potencjał elektrody chlorosrebrowej wynosi 0, 34 V
Inaczej: Jeżeli podane jest E’Ag+/Ag = 0, 80 V to wtedy Potencjał elektrody srebrowej wyraża się wzorem: = 0, 80 V + 0, 059 log a. Ag+ Ponieważ Ks. Ag. Cl=1, 6 10 -10 = [Ag+] [Cl-] , więc [Ag+] = 1, 6 10 -10 / 0, 01 = 0, 80+ 0, 059 log 1, 6 10 -10 - 0, 059 log 0, 010 = 0, 222 - 0, 059 log 0, 010 = 0, 34 V ( Na czerwono normalny potencjał elektrody chlorosrebrowej względem NEW) Odp. Potencjał elektrody chlorosrebrowej względem NEW wynosi + 0, 340 V
• Przykład 2. Obliczyć potencjał redoks układu 0, 3% (m/m) roztwór Na. I+stały Ag. I, jeżeli Ks. Ag. I=2 10 -16. E’ 0 Ag+/Ag wynosi + 0, 80 V, temp. 25 o. C (Na – 23; I – 127; Ag – 108). Mamy tutaj do czynienia z elektrodą drugiego rodzaju, elektrodą jodosrebrową, jest to więc przypadek analogiczny do poprzedniego, tylko Ks jest inny. Schemat elektrochemiczny tej elektrody Ag/ Ag. I(s) /I-. Przemiany zachodzące w elektrodzie można zapisać: Ag + I- Ag. I + 1ē wzór na potencjał
Aby obliczyć stężenie [I-], należy uwzględnić fakt, że Na. I Na+ + I-: n Na. I = n Icm Na. I = [I-] = 0, 02 mol /L
Ponieważ Ks. Ag. I l= 2 10 -16 = [Ag+] [ I-] , więc [Ag+] = 2 10 -16 / 0, 02 E’ = + 0, 80 V + 0, 059 log Ks. Ag. I – 0, 059 log [I-] Normalny potencjał elektrody jodosrebrowej = – 0, 059 log [I-] E’ = + 0, 80 V + 0, 059 log 2 10 -16 – 0, 059 log 0, 02= 0, 1260+0, 1 = -0, 026 V Normalny potencjał elektrody jodosrebrowej Odp. Potencjał układu Ag(Ag. I(s)/Na. I wynosi – 0, 026 V.