Le tecniche cromatografiche permettono la separazione e quantificazione















: tempo necessario alla")


 della cromatografia per scambio")







 che viene digitalizzato")


")






 vs. Split (100: 1) Syringe Injector He He Purge valve closed")
 flowing at 1 m. L/min 0. 32")














- Slides: 54
Le tecniche cromatografiche permettono la separazione e quantificazione dei componenti di matrici complesse e trovano quindi numerosissime applicazioni in campo alimentare. Nei metodi cromatografici i componenti di una miscela si separano distribuendosi tra due fasi: una fase stazionaria (un solido o un liquido su supporto solido inerte) una fase mobile (gas o liquido) che fluisce in modo continuo su quella stazionaria. La separazione è dovuta principalmente alle relative affinità per la fase stazionaria. Nella cromatografia liquida (LC) la fase mobile è un liquido, nella gas cromatografia (GC) la fase mobile è un gas. 14
CLASSIFICAZIONE DEI METODI CROMATOGRAFICI 15
PRINCIPI TEORICI DELLA CROMATOGRAFIA Esempio: separazione di un campione a tre componenti in una colonna chiusa. La fase stazionaria consiste di particelle solide porose contenute all’interno di un tubo lungo e sottile (colonna). Nel passaggio attraverso la colonna ogni componente X si distribuisce fra la fase stazionaria (s) e la mobile (m): Xm Xs Il coefficiente di ripartizione (o di distribuzione) del componente X è definito come: A: il campione viene iniettato all’entrata della colonna B D: la fase mobile fa spostare il campione attraverso la fase stazionaria p. 518 Flusso del solvente Quale componente ha K maggiore? A B C D 16
t. R t. M p. 521 Tempo di ritenzione (t. R): tempo necessario alla sostanza iniettata per essere eluita dall’inizio all’uscita della colonna. Tempo morto (t. M): tempo di ritenzione di un composto che non è trattenuto e che passa attraverso la colonna alla stessa velocità con cui fluisce la fase mobile lungo la colonna. Analogamente si definiscono i corrispondenti: Volume di ritenzione (VR): volume di fase mobile necessario ad eluire l’analita dall’inizio all’uscita della colonna. Volume morto (VM): il volume di ritenzione di un composto che non è trattenuto (corrisponde al volume di fase mobile che occupa la colonna). 17
Si può dimostrare che: Effetto della selettività, dell’efficienza e del fattore di capacità sulla risoluzione scarsa picchi non separati buona risoluzione dovuta a buona efficienza picchi stretti buona risoluzione dovuta a buona selettività picchi distanti risoluzione scarsa dovuta ad un basso fattore di capacità 18
Le applicazioni della cromatografia vanno dall’analisi qualitativa a quella quantitativa di miscele anche molto complesse. L’analisi quantitativa sfrutta la misurazione dell’altezza o dell’area dei picchi. La misurazione dell’area è più affidabile in quanto non risente dell’eventuale allargamento dei picchi in seguito a variazione delle condizioni di lavoro. I metodi di analisi sono tutti indiretti. Si costruisce prima una curva di calibrazione per ciascun analita e poi si ricava la concentrazione dell’analita nella miscela in esame mediante interpolazione. Il metodo dello standard interno è il più affidabile: una quantità nota di standard viene introdotta nelle soluzioni standard e nel campione in esame; il parametro analitico è quindi costituito dal rapporto tra le aree dello standard e dell’analita (il metodo funziona solo se il picco dello standard è vicino ma separato da quello dell’analita). 19
Cromatografia Ionica | È la variante ad alta pressione (prestazioni) della cromatografia per scambio ionico | Serve per separare e quantificare ioni, in particolare inorganici, utilizzando resine a scambio ionico
Il cromatografo ionico è sostanzialmente un HPLC ed è composto da: Riserva di eluente Valvola Iniezione Pompa tipo HPLC Colonna Soppressore Integratore Rivelatore a Conducibilità
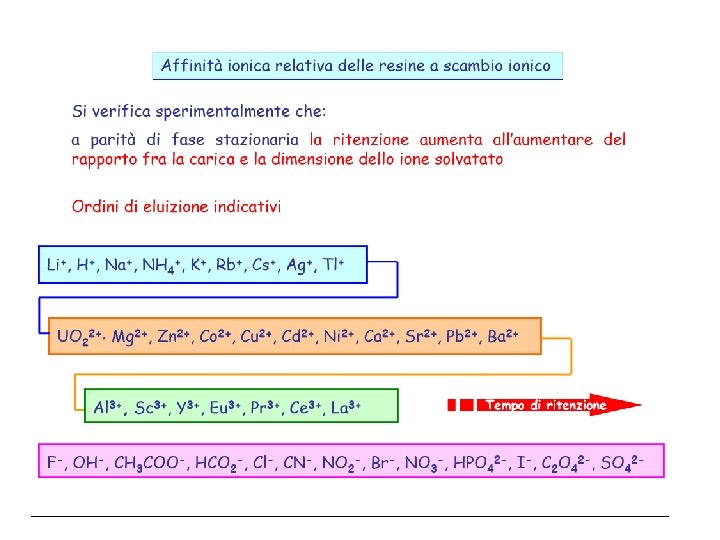
Cromatografia per Scambio Ionico Nella cromatografia per scambio ionico, la ritenzione si basa sull’attrazione tra gli ioni del soluto e i siti carichi legati alla fase stazionaria. La fase stazionaria è costituita da una resina a scambio ionico, ossia un supporto organico al quale sono legati dei gruppi ionici o ionizzabili (ad esempio –SO 3 -, -COO-, NR 3+). I gruppi ionici della resina trattengono per mezzo di interazioni elettrostatiche, controioni di carica opposta che possono essere scambiati con gli ioni presenti nella fase mobile. Il meccanismo di separazione è basato perciò sulla diversa affinità che i diversi soluti (ionici) presentano nei confronti dei gruppi attivi della resina.
Colonne Sono il cuore dello strumento. La separazione avviene per una serie di equilibri di scambio tra fase stazionaria e fase mobile (eluente) Gli ioni con carica netta più elevata (al p. H dell’eluente) vengono trattenuti più fortemente A parità di carica vi è un ordine di selettività che dipende dal tipo di fase stazionaria (dipende dalle costanti di scambio) Forti Deboli
Ampio intervallo di p. H di utilizzo La comune definizione delle resine ioniche è fatte sulla base della acidità del loro gruppo attivo Cationico Anionico Forte -SO 3 - H+ -N+(CH 3)3 OH- Debole -COO- H+ -N+H 3 OH- Ridotto intervallo di p. H di utilizzo
Eluente La fase eluente deve contenere elettroliti forti, composti ionici completamente dissociati, in grado di competere con gli analiti nello scambio con la resina. Gli analiti vengono eluiti più velocemente quanto più è alta la concentrazione di competitori. Può contenere solventi organici (CH 3 CN, Me. OH, ecc. ) per evitare l’adsorbimento per interazione idrofobica su resina o gruppi funzionali di analiti organici Se rimane costante durante tutta l’analisi -> Eluizione Isocratica Se cambia di composizione durante l’analisi -> Gradiente di Eluizione Forza Ionica p. H Concentrazione dei costituenti Il gradiente si può comporre: | sotto pressione con una pompa per ogni componente (più riproducibile ma strumentazione più costosa) | non sotto pressione con l’ausilio di un miscelatore posto prima della pompa
Rivelatore Dato che si determinano ioni, il rivelatore universalmente utilizzato è costituito da una piccola cella conduttimetrica. Applicando una piccola ddp tra i due elettrodi, fluisce una corrente proporzionale alla conducibilità dell’eluente + analita. Problema: poiché i rivelatori a conducibilità rispondono a tutti gli ioni e l’eluente è costituito da una soluzione elettrolitica concentrata, il suo segnale coprirebbe quello degli analiti. Come è possibile ovviare al problema?
La conducibilità dell’eluente viene eliminata mediante un Soppressore L’unità di soppressione può essere costituita da: | Cartucce usa e getta | Micromembrana rigenerata controcorrente | Micromembrana rigenerata elettroliticamente Il soppressore è uno scambiatore di ioni di segno opposto rispetto agli analiti ed ha lo scopo di diminuire il segnale di fondo dato dagli ioni contenuti nell’eluente.
Integrazione e quantificazione Il rivelatore produce un segnale elettrico analogico (E) che viene digitalizzato e “contato” tramite un integratore (o un interfaccia A/D e un computer). L’area dei picchi del cromatogramma è proporzionale alla concentrazione dell’analita rendendo possibile la quantificazione.
Separazione di Anioni es. Cl-, NO 3 - e SO 42 - La configurazione dello strumento è: colonna con resine ammonio quaternario in forma HCO 3 -; soppressore in forma H+; rivelatore conduttimetrico. Gli analiti in esame sono dissociati a qualsiasi p. H quindi scegliamo come eluente una miscela Na 2 CO 3/Na. HCO 3 Nella colonna gli anioni si separano ed escono a diversi tempi in forma Na+ (Na+Cl-, …) insieme ad un largo eccesso di Na+HCO 3 - e Na+2 CO 32 - Tutti gli analiti, trasformati in acidi forti, (H+Cl-, …) danno un segnale mentre l’eluente, ora H 2 CO 3 poco dissociato, non viene rivelato Nel soppressore il catione coordinato (Na+) viene sostituito da H+.
Separazione di Cationi es. Na+, K+ e NH 4+ La configurazione dello strumento è: colonna con resine solfoniche in forma H+; soppressore in forma OH-; rivelatore conduttimetrico. In questo caso l’eluente è HCl Nella colonna i cationi competono con H+ e si separano. Escono coordinati a Cl - (Na+Cl-, …) insieme ad un largo eccesso di H+Cl- Tutti gli analiti, trasformati in idrossidi dissociati, (Na+OH-, …) danno un segnale Nel soppressore l’anione coordinato (Cl-) viene sostituito da OH- e l’eccesso di H+ viene neutralizzato ad H 2 O.
GASCROMATOGRAFIA La fase mobile è un gas. Le sostanze da separare (liquidi, solidi, gas) devono essere portate ad una temperatura sufficiente a renderle gassose o comunque portarle allo stato di vapore. Cromatografia di adsorbimento gas-solido (fase stazionaria = solido adsorbente) Cromatografia di ripartizione gas-liquido (fase stazionaria = liquido che può essere supportato da un solido inerte o depositato sulle pareti della colonna) A differenza della LC, la fase mobile non ha effetto competitivo: il gas di trasporto serve solo per trascinare i componenti lungo la colonna.
SCHEMA DI UN GASCROMATOGRAFO He, Ar, N 2, H 2 33
Iniezione ed iniettori Il campione viene iniettato in quantità molto piccole (fino a 0, 5 ml per impaccate, fino a 100 volte inf. per capillari). L’entrata deve essere ad una temperatura sufficientemente elevata da permettere l’evaporazione istantanea del campione e abbastanza grande da permettere al vapore di espandersi senza essere spinto indietro. Colonne gascromatografiche Si suddividono in impaccate e capillari. Le impaccate contengono particelle solide che vengono impaccate: diatomee o diatomee silanizzate, per ridurre la polarità mediante arricchimento superficiale di gruppi metilici. Le capillari sono tubi molto più sottili e lunghi (diametro = 0. 3 -0. 5 mm; lunghezza = 50 -300 m) in cui la fase stazionaria è depositata sulle pareti interne. Queste ultime hanno un elevato numero di piatti teorici (anche 300. 000) grazie alla loro elevata lunghezza. 34
Colonna capillare standard a fase legata 35
45° 145° Influenza della T sui t. R: più la temperatura è elevata più il soluto tende a trasferirsi nella fase gassosa aumentando T diminuisce t. R programma di T 36
Se l’analisi riguarda la quantificazione di cationi, o anioni, in un acqua di scarico, potrebbe convenire ricorrere alla cromatografia ionica. 37
Function • Separation of volatile organic compounds • Volatile – when heated, VOCs undergo a phase transition into intact gas-phase species • Separation occurs as a result of unique equilibria established between the solutes and the stationary phase (the GC column) • An inert carrier gas carries the solutes through the column 38
Splitless (100: 90) vs. Split (100: 1) Syringe Injector He He Purge valve closed GC column Purge valve open 39
Open Tubular Capillary Column Mobile phase (Helium) flowing at 1 m. L/min 0. 32 mm ID Liquid Stationary phase 0. 1 -5 mm 15 -60 m in length 40
41
Who Discovered SPME? • Solid Phase Microextraction was invented in 1990 by Dr. Janusz Pawliszyn and his colleagues from the University of Waterloo in Canada. • He invented this technique to “address the need for a fast, solventfree, and field compatible sample preparation method”, which faster and more efficient is the name of the game in industry.
What is an SPME? • SPME, also known as “Spee Mee”, is a solvent-free adsorption/desorption technique. • It consists of coated fibers that are used to isolate and concentrate analytes into a range of coating materials. • After extraction, the fibers are transferred to an analytical instrument for separation and quantification of the target analytes. • This is accomplished with the help of a syringe-like handling device that protects your sample while transferring from your sample to the instrument. • This syringe-like device also protects your fiber during storage.
How does SPME work? • • • First, you draw the fiber into the needle. The needle is then passed through the septum that seals the vial. You then depress the plunger to expose the fiber to your sample or headspace above the sample. Organic analytes are then adsorbed to the coating on the fiber. After adsorption equilibrium is attained, which can be anywhere from 2 minutes to 1. 5 hours, the fiber is drawn back into the needle and is withdrawn from the sample vial. Finally, the needle is introduced into the GC injector or SPME/HPLC interface, where adsorbed analytes are thermally desorbed and delivered to the instruments column.
Components of a Manual SPME Holder Adjustable needle guide/depth gauge Plunger The O ring Plain Hub Septum piercing needle Where fiber is exposed in headspace/liquid sample Plunger retaining Screw SPME manual holder
SPME fibers available • Fiber coating available: – – – – PDMS/DVB Polyacrylate CAR/PDMS CW/DVB CW/TPR Stable. Flex DVB/CAR/PDMS • Different Phases available: – Non-bonded • stable w/ some water-miscible organic solvents • slight swelling may occur • NEVER use nonpolar organic solvents – Bonded • stable with ALL organic solvents • slight swelling possible w/ nonpolar solvents – Partially Crosslinked • stable in most water-miscible organic solvents • May be stable in some nonpolar solvents, but slight swelling possible – Highly Crosslinked • Equivalent to the partially crosslinked, but some bonding to core has occurred in the past
Recommended Temperature and Conditioning for GC Use Maximum Operating Conditioning Time Phase Thickness Temperature (Hrs. ) PDMS 100μm 280°C 200°C-270°C 250°C 1 30μm 280°C 200°C-270°C 250°C 1 7μm 340°C 220°C-320°C 2 -4 PDMS/DVB 65μm 270°C 200°C-270°C 260°C 0. 5 Polyacrylate 85μm 320°C 220°C-310°C 300°C 2 CAR/PDMS 75μm 320°C 240°C-300°C 280°C 0. 5 CW/DVB 65μm 265°C 200°C-260°C 250°C 0. 5 DVB/CAR/PDMS 50/30μm 270°C 230°C-270°C 4 Note that the Polyacrylate, or white fiber, will turn brown as a result of condition and will not hurt the performance of the fiber.
Injecting and Running a Sample on GC This is where you inject your SPME needle on the GC-MS
Different fields using SPME • Applications SPME is applied to include: – Food and drug – Environmental – Clinical/Forensics
• Environmental Application Air sample – Analytes are extracted by the fiber wither by direct exposure or by use of the headspace method. – Most applications involve the use of a commercial SPME fiber, but a dialuminum trioxide-coated fiber has been – – • Water samples – Can be performed by direct immerion (DI), headspace (HS), or in-tube method. – The air inside needle must be completely replaced by water and effects of extracted analytes on the external – – – • used for VOC sampling. On-site air sampling can be performed by the equilibrium methods or by the non-equalibrium method, with quantification by use of calibration plots from a standard gas generating system of standard gas mixture as opposed to using equations. rapid air sampling can be performed with controlled air-flow rate and quantified by use of diffusion-based calibration methods by use of wither the interface or cross-flow model. wall of the needle should be avoided. The in-tube SPME has been used for analysis of BTEX, PAH, pesticides, and herbicides in aqueous samples. The fibers have also been used to analyze environmental pollutants in aqueous samples and have been accompanied by ultrasounds or microwaves. Traditional calibration methods have been used for most applications, but diffusion-based calibration methods have been used. Soil and sediment samples – Performed by HS or DI methods and applications have been assisted by sonication, microwaves or by heater or – – cooling fiber. Traditional calibrations but some exhaustive calibration methods have been used in quantification of BTEX in soil samples. A hollow-fiber membrane-protected SPME has also been used for determination of herbicides in sewage-sludge samples.
List of Environmental Applications -gas
List of Environmental Applications-aqueous
List of Environmental Applications-soil/sediment