Evolution of Plasmodium falciparum var antigen genes Przeworski

















































- Slides: 51
Evolution of Plasmodium falciparum var antigen genes Przeworski lab meeting
Evolution of Plasmodium falciparum var genes § Malaria: a protozoan blood parasite carried by mosquito vectors principally of the genus Anopheles § Plasmodium falciparum and Plasmodium vivax are the main species infectious to humans § Related species infect primates, rodents, other mammals, birds and reptiles § P. falciparum is the most deadly: in humans it is responsible for § 80% of infections (250 million annually) § 90% of deaths (1 million annually) § Endemic in tropical and sub-tropical regions § Member of the Apicomplexa, a phylum characterized by a unique organelle called the apical complex/apicoplast responsible for lipid synthesis
Evolution of Plasmodium falciparum var genes § Key elements of life cycle: § § Infection with diploid cells from mosquito bite Asexual replication in liver and then red blood cells Haploid cells produced in red blood cells, ingested by mosquito Sex in mosquito produces diploid cells that migrate to saliavary gland § Cycles of replication and aggregation in blood vessels (sequestration) causes the symptoms of malaria: § Anaemia § Fever, chills, malaise § Coma (especially in cerebral malaria) § Sequestration prevents clearing of infected red blood cells by the spleen
Evolution of Plasmodium falciparum var genes § The var genes are responsible for antigenic variation and sequestering by cytoadhesion § They encode Pf. EMP-1 (P. falciparum erythrocyte membrane protein), a parasite protein exported to the outside of the host red blood cells § Typically 60 var genes per genome, clustered telomerically. Differential expression allows immune evasion § Protein consists of Duffy binding-like domain (DBL) and cysteine-rich interdomain region (CIDR) § All proteins begin with DBL 1 , hence its use in population biology Kraemer et al. 2007 BMC Genomics 8: 45
Evolution of Plasmodium falciparum var genes § Collaboration with Caroline Buckee (Oxford, Santa Fe and Kilifi) and Pete Bull (Kilifi) § Looking at 1000 DBL sequences § Aims § Investigate genetic structuring imposed by geography, selection and genomic position § Understand population biology: evidence for strain structure? § Reconstruct evolutionary history: quantify the roles of gene duplication, homologous and non-homologous recombination § Current hypotheses: § Categorization based on conserved 5’ regions and double-W § Recombination hierarchy § Some genotypes are over-represented in severe malaria
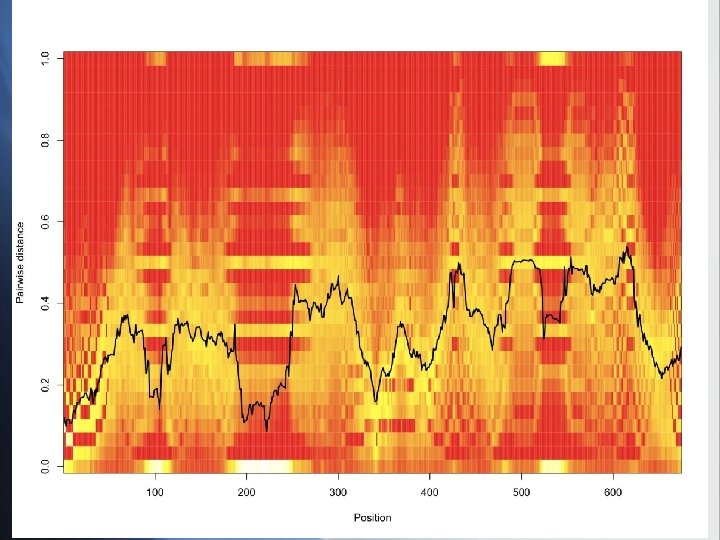
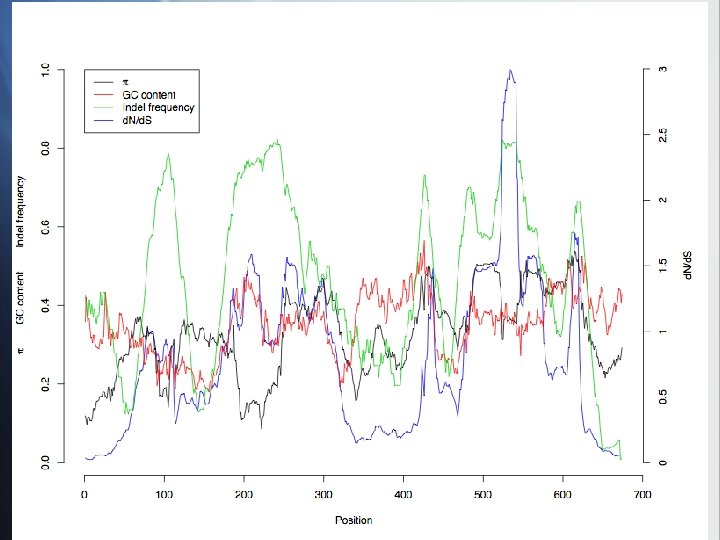
var genes: raw data
Evolution of Plasmodium falciparum var genes § Alignment is a major problem even in the conserved DBL domain § Multiple alignment algorithms § Clustal (Larkin et al. 2007 Bioinformatics 23: 2947 -8) § Muscle (Edgar 2004 Nucleic Acids Research 32: 1792 -7) § T-Coffee (Notredame et al. 2000 JMB 302: 205 -17) § Pairwise alignment § BLAST (Tatiana et al. 1999 FEMS Microbiol Lett 174: 247 -250) § Hidden Markov models (e. g. Lunter 2007 Bioinformatics 23: 2485 -7) § Multiple statistical alignment § Phylogenetic (Holmes 2003 Bioinformatics S 1: i 147 -57)
Malign: probabilistic sequence alignment in malaria as a pre-requisite for evolutionary inference § Simulating a sequence alignment § The likelihood of a sequence alignment § Bayesian inference § Testing, testing § Inference proper
Simulating an alignment: beta-binomial distribution § N=5 sequences, T=1 site § Simulate the frequency of indels from a symmetric beta distribution with parameters ( , ) § Given the frequency f, draw the number of indels from a binomial distribution with parameters (N, f)
Simulating an alignment: beta-binomial distribution § For example, the beta-binomial distribution with N=15 and § =0. 1 (red) § =1 (yellow) § =10 (green) § The binomial distribution with p=0. 5 (blue)
Simulating an alignment: beta-binomial distribution
Simulating nucleotides: multinomial-Dirichlet distribution § Simulate the nucleotide frequencies from a symmetric Dirichlet distribution with parameter =( , , , ) § Given the frequencies f=(f. A, f. G, f. C, f. T), simulate the number of As Gs Cs and Ts from a multinomial distribution with parameters (N, f) = 0. 1 = 10
Simulating nucleotides: multinomial-Dirichlet distribution
Simulating a nucleotide alignment T = 100 L = 77
Simulating a nucleotide alignment True alignment Raw data
Representing an alignment § Matrix of size N by L § 0 indicates an indel § Other integers correspond to the position in the raw sequence
The likelihood of an alignment: indel pattern § Let I be the number of indels, and be the parameter of the beta distribution. N is the number of sequences. § For a single column in the alignment, the beta-binomial distribution gives the probability of I § However, the sequences are labelled, so the ordering matters. Therefore the likelihood is
The likelihood of an alignment: nucleotide pattern § Let Yi, i={A, G, C, T} be the number of each type of nucleotide in the remaining (N-I) non-indels. Let be the parameter of the Dirichlet distribution. § For a single column, conditional on the indel pattern, the multinomial-Dirichlet distribution gives the likelihood of Y § Again the ordering matters, so the likelihood is
The likelihood of an alignment: missing columns § Because our representation of the alignment does not specify the exact position of columns fixed for indels, we need to take that into account in the likelihood. § When there are (T-L) columns fixed for indels, their likelihood is
The full likelihood of an alignment
Review of assumptions § Independence between sites = free recombination § Exchangeability between sequences = panmictic population structure § Equal base usage and stationary frequency distribution § Simplistic indel model
Bayesian inference § Fix the auxiliary variables, e. g. at N=15 and T=100 § Priors on and , e. g. exponential distribution with mean 0. 1 § Markov chain Monte Carlo (MCMC) for sampling from the joint posterior distribution of , and the alignment
MCMC: Multiplicative updates to and § E. g. propose an update from to ´: § Let U ~ Uniform(-1, 1) § Let ´ = exp(U) § The acceptance probability is
MCMC: Updating the alignment § General strategy is to partition the current alignment at random
MCMC: Updating the alignment § And re-align one partition relative to the other
MCMC: Updating the alignment § Gaps are stripped from both
MCMC: Updating the alignment 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 § And the reference partition (top) is labelled § Odd-numbered sites correspond to gaps, even-numbered sites to matches
MCMC: Updating the alignment 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 § Each site in the non-reference partition is aligned to a numbered site in the reference partition
MCMC: Updating the alignment 0 1 2 3 4 0 0 1 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 7 11 15 17 18 § Gaps may have multiple hits, but matches cannot 21
MCMC: Updating the alignment § Finally the alignment is reconstructed
MCMC: Updating the alignment § In this example, it has grown by two columns
MCMC: Updating the alignment Partitioning the alignment probabilistic Stripping indels book-keeping 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Realigning the partitions probabilistic 0 0 1 7 11 15 17 18 21 Reconstructing the alignment book-keeping
MCMC: Partitioning the alignment § The partition size is drawn from some discrete distribution between 1 and N-1, e. g. uniform, or inverse. § Sequences are then assigned to one partition or the other uniformly at random (formally, the hypergeometric distribution describes this method of sampling without replacement). § The proposal probability is independent of the alignment, so the ratio of reverse and forward proposal probabilities (the Hastings ratio) is 1.
MCMC: Realigning the partitions 0 1 2 3 4 0 0 1 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 7 11 15 17 18 21 § In principal, you could enumerate over all possible alignments of the non-reference to the reference partition. (Very approximately (2 L)L combinations). § For each combination you could calculate the posterior probability and Gibbs sample. § In practice, not computationally tractable so I impose a window of, e. g. ± 40 matches, which constrains the maximum distance a site in the nonreference partition can move from its current position. § The likelihood can be calculated it an efficient manner (computer scientists would call it dynamic programming).
MCMC: Realigning the partitions 0 1 2 3 4 0 0 1 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 7 11 15 17 18 21 § Since not all possible moves are allowed, the proposal is a Metropolis. Hastings move. § To calculate the reverse proposal probability for the Hastings ratio, it is necessary to implement the move and carry out the full computations a second time. § The proposed change in the alignment, A´, is accepted with the usual probability
R/C implementation § The MCMC code is written in C for speed § Linked as a static library and called from R for graphical monitoring and post-processing § Can watch the chain and the current alignment in real time
Testing, testing § It is criminally insane to assume your MCMC will work (straight away) § Two methods of testing are highly recommended § Do you retrieve the prior when there is no data? § Is the posterior well-calibrated?
Do you retrieve the prior when there is no data? § Testing the prior for alpha and beta is straightforward: you feed the program no data and compare to the specified prior distribution. § How do you test the “prior” on the alignment? § The “prior” on the alignment is conditional on the sequence lengths but not on the nucleotide sequences themselves § Calculate it theoretically for sufficiently simple examples § Use an alternative method for sampling from the prior, such as importance sampling, and compare.
Testing the alignment prior: theoretical approach § For the 2 -sequence case, this is theoretically tractable. Suppose both sequences have length S, and the maximum alignment length is T. Calculate the alignment length Pr(L|S). Configuration Count 2 S-L L-S T-L Prior probability
Testing the alignment prior: theoretical approach § E. g. T=100, S=50, =0. 1 § Key § MCMC: black lines § Theory: red lines
Testing the alignment prior: importance sampling § Importance sampling is an alternative to MCMC. § Instead of proposing local moves to the parameters and missing data, they are independently proposed de novo each iteration. § Each simulation is given a weighting equal to the ratio of the posterior and proposal probabilities. § The proposal distribution is chosen to approximate the posterior distribution, and thus minimize the variance in the weightings. § I used a sequential importance sampler (SIS) that simulates the first sequence, then the second given the first, and so on. § Each sequence is simulated to ensure agreement with the observed sequence length. § The importance sampler represents a better choice for simulating from the prior, but not necessarily the posterior.
Testing the alignment prior: importance sampling § E. g. N=15, T=100, =0. 3 S=[51, 52, 47, 57, 47, 50, 53, 48, 49, 44, 47, 52, 50] § Key § MCMC: white bars 3000 iterations § SIS: grey bars 1000 iterations
Is the posterior well-calibrated? § The well-calibrated Bayesian is a person whose prior beliefs reflect the long-run probabilities of an event. § Dawid, A. P. (1982) JASA 77: 605 -10 § For example, if a weather forecaster predicts the chance of rain each day, s/he is well calibrated if, in the long-run, it rained on at least 80% of the days when the predicted chance of rain was 80% or more. § This is a minimum requirement for good inference: a lazy weather forecaster would be well-calibrated if s/he used the annual precipitation rate as his/her estimated chance of rain every day.
Is the posterior well-calibrated? § The idea of calibration can be used to test a Bayesian inference procedure, using the following simulation scheme § Simulate a parameter from a prior p( ) § Simulate data X from a model p(X| ) § Perform inference using the same prior and likelihood to obtain a posterior distribution phat( |X) § Calculate a 95% credible interval based on phat( |X) § Repeat many times § If the method of inference is working correctly, the credible intervals must be well-calibrated. § A calibration curve shows the proportion of simulations for which the true value of fell within the 100(1 - )% credible interval. § To be well-calibrated means that proportion equals 1 -
Is the posterior well-calibrated?
Debugging § Check the likelihood calculations are consistent § For accepted moves, check the calculated reverse move probability was correct § Go through the calculations for simple examples by hand § Theory - go through again by hand
Inference proper § Screw debugging, let’s analyse the data § What will we do with a posterior distribution of sequence alignments? § For inference § Improper inverse priors on and § Improper uniform distribution on the maximum alignment length T
Conclusions § There was a bug in my MCMC § Multiple sequence alignment allows § Exploratory analysis of quantities of interest pertaining to selection, recombination and demography § Formal model fitting using standard tools such as omega. Map, LDhat and Structure-like analyses