DELECIONI SINDROMI CRIduCHAT SY Leenov sindrom 5 p

; 5 p- Opisali su Lejeune i sar. 1963. Delecija malog")
.")
. - 5 p 15.")






 i Angelmanov sindrom Prader-Willi i Angelman sindromi nastaju delecijom (brisanjem)")

 nasleđenom")
, ili")
- Slides: 16
DELECIONI SINDROMI
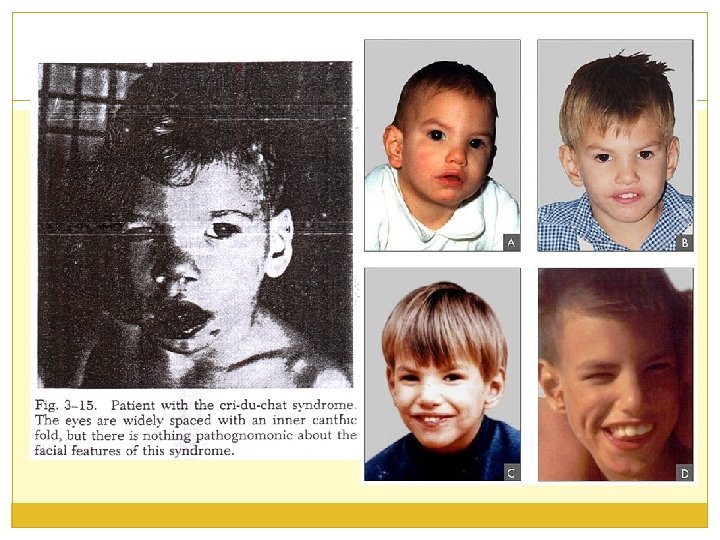
CRI-du-CHAT SY (Leženov sindrom); 5 p- Opisali su Lejeune i sar. 1963. Delecija malog segmenta 5 p (del 5 p ili 5 p - ). Incidenca sy u opštoj populaciji je 1: 30 000 -50 000. Češće su zahvaćene devojčice. Sindrom je dobio naziv prema plaču ove dece koji je nalik mjaukanju mačke.
Klinička slika Novorođenčad su male PTT, PTD, mikrocefalična. Hipotonija (vremenom može preći u hipertoniju). Lice okruglo (moon face), epikantus, antimongoloidni položaj očiju, hipertelorizam, ugnut koren nosa. Zbog anomalija larinksa plač deteta je nalik mjaukanju mačke (5 p 15. 3). 20% ima USM, anomalije CNS-a, bubrega, kostiju. PMR je usporen, retardacija rasta. Teška mentalna zaostalost, IQ je manji od 20. Poremećaj ponašanja - hiperaktivni, agresivni, stereotipni pokreti. Dužina života ove dece je varijabilna.
Citogenetika Najčešće je posledica de novo mutacije – delecije (90%). - 5 p 15. 2, region odgovoran za kliničku sliku sindroma (gen CTNND 2 se povezuje sa umnom zaostalošću) - 5 p 15. 3, region odgovoran za plač nalik mjaukanju mačke Slučajevi porodične translokacije ili ring hromozoma ipak nisu tako retki pa je PND potrebna u narednim trudnoćama.
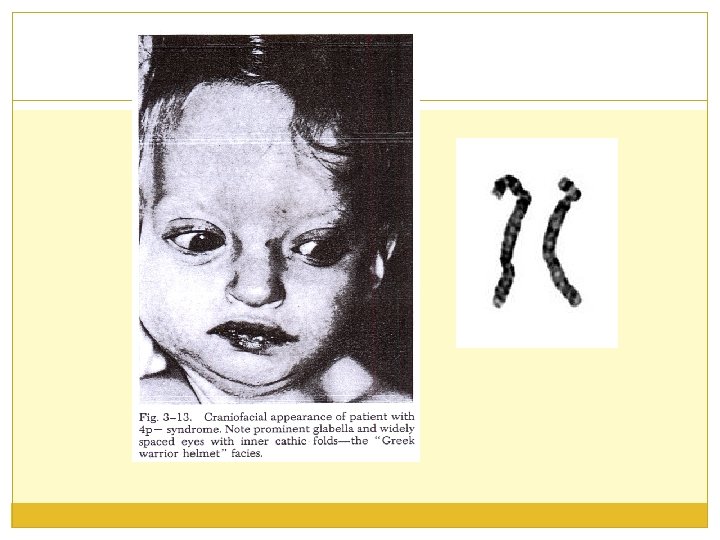
Wolf sy ; 4 p- Icidenca sindroma je 1: 50 000. Delecija u nivou 4 p 16. 3 dovodi do kliničke slike ovog sy. Dva puta češće su zahvaćene devojčice. IUZ u rastu i razvoju je veoma izražen. Česti su spontani pobačaji i mrtvorođenost.
Klinička slika Novorođenčad su male PTT, PTD, hipotonična. Mikrocefalija, izbočeno čelo, hipertelorizam, ptoza kapaka, epikantus, nistagmus, strabizam. Nos je širok i kratak, uši velike, loše modelirane, nisko postavljene, suženog sprovodnog kanala. Usne okrenute na dole, rascep usne i nepca. Vrat kratak, trup izdužen, prisutna prekobrojna rebra. Klinodaktilija 5. prsta šake, anomalije stopala, brazda 4 prsta, ostali dermatoglifi hipoplastični. USM (40%), anomalije CNS-a, digestivnog i urogenitalnog sistema. PMR je voma usporen, IQ je oko 20. Dužina života je varijabilna (opisan je bolesnik sa 24 godine).
citogenetika U 90% slučajeva sindrom je de novo mutacija. U 10% slučajeva povezan je sa roditeljskim mozaicizmom ili translokacijom. Geni koji se dovode u vezu sa sy: NSD 2 – karakterističan izgled lica. LETM 1 - povezan sa epi napadima. MSX 1 - abnormalnost zuba, rascep usne i nepca. Genetski savet- u narednim trudnoćama potrebna je PND.
Williams sy Williamsov sindrom je posledica delecije oko 26 -28 gena na 7 hr. ( del 7 q 11. 23). Sindrom je 1961. opisao John P. C. Williams. Učestalost sindroma je 1: 7. 500 – 10. 000. Citogenetika: Najčešće je de novo delecija (greška nastala tokom oogeneze ili spermatogeneze). U veoma malom procentu javlja se kod zahvaćenih roditelja (obolelih), po tipu autozomno dominantnog oblika nasleđivanja.
Klinička slika Preko 60% ima USM, hiperkalcemija, hipotireoza. Ova deca imaju usporen PMR. Imaju specifičan izgled lica sa jako izraženim spojenim obrvama, zadebljanje periorbitalne i perioralne regije, ugnut koren nosa, hipertelorizam, nistagmus, strabizam, loše modelirane ušne školjke, hiperakuzu. Problemi motoričke prirode, koordinacije, hipertonija, hiperrefleksija. Prisutni su umerena mentalna retardacija i problemi vezani za kognitivne funkcije i socijalnu adaptaciju. Približno 10% osoba sa Williamsovim sindromom ima znake autizma.
Prader Willi sindrom (PWS) i Angelmanov sindrom Prader-Willi i Angelman sindromi nastaju delecijom (brisanjem) iste regije hromosoma 15 (q 11 -q 13). PWS i Angelmanov sindrom imaju posebnu važnost kao prvi dokaz genomskog imprintinga. Genomski imprinting - je fenomen u kome aktivnost gena zavisi od roditeljskog porekla hromozoma. Imprintirani gen je ispoljen samo s jednog roditeljskog alela, dok je alel nasleđen od drugog roditelja utišan (inaktiviran). Ovakva ekspresija je u suprotnosti sa mendelovim nasleđivanjem kod koga oba alela jednako doprinose fenotipu.
Kod osoba koje imaju funkcionalne majčine gene te regije (tj. očeva kopija gena je “utišana”), i ako nastane delecija u tom regionu - razvija se Angelmanov sindrom; dok kod osobe koja ima funkcionalne očeve gene (majčina kopija je “utišana) i dođe do delecije gena na očevom hromozomu, razvija se PWS. Učestаlost ovа dvа sindromа je 1: 10 000 do 1: 25 000. Više genetičkih modela učestvuje u nastajanju sindroma, a mehanizam nasleđivanja odstupa od Mendelovog. Genetička dijagnoza se postavlja molekularno – genetičkim metodama.
Prader-Willi-jev sindrom Nаstаje kаo posledicа delecije gena na 15. hromosomu (q 11 -13) nasleđenom od oca (75%); ili je u pitanju uniparentalna dizomija hromozoma 15 (oba hromozoma su poreklom majke, 25%). Klinička slika: Hipotonija, nizak rast, blaga mentаlna zaostalost, poremećаj u ishrаni - nekontrolisаni аpetit (hiperfagija), hipogonadizam (sterilitet), dijаbetes. Jedan od većih problema je morbidna debljina zbog stalnog osjećaja gladi. Lečenje : hormon rasta, fizikalna terapija, govorna i radna terapija.
Angelman-ov sindrom Nаstаje kаo posledicа delecije hromozomа 15 koji potiče od mаjke (mаternаlni), ili je u pitanju uniparentalna dizomija hromozoma 15 (oba hromozoma su poreklom oca). Klinička slika: teška psihomotorna zaostalost, epilepsija, ataksija, kаo i rаzličite mаnifestаcije u ponаšаnju – hiperaktivni su, nedostatak pažnje, često se smenjuju fаze smehа, poteškoće sa spavanjem. Lečenje: antikonvulzivi, fizikalna terapija, govorna i radna terapija.