Catabolism of the carbon skeletons of amino acids

Catabolism of the carbon skeletons of amino acids The catabolism of the amino acids found in proteins involves : �the removal of α-amino groups. � followed by the breakdown of the resulting carbon skeletons. These pathways converge to form seven intermediate products : oxaloacetate, α-ketoglutarate, pyruvate, fumarate , succinyl Co. A, acetyl Co. A, and acetoacetyl Co. A. These products directly enter the pathways of intermediary metabolism, resulting: �in the synthesis of glucose or lipid. � or in the production of energy through their oxidation to CO 2 and water by the citric acid cycle.

Transamination typically initiates amino acid catabolism Removal of -amino nitrogen by transamination is the first catabolic reaction of amino acids except for proline, hydroxyproline, threonine, and lysine. The hydrocarbon skeleton that remains is then degraded to amphibolic intermediates as outlined in Figure 1.

Glucogenic and ketogenic amino acids Amino acids can be classified as glucogenic or ketogenic based on which of the seven intermediates are produced during their catabolism. A. Glucogenic amino acids Amino acids whose catabolism yields pyruvate or one of the intermediates of the citric acid cycle. These intermediates are substrates for gluconeogenesis and, therefore, can give rise to the net formation of glucose or glycogen in the liver and glycogen in the muscle. B. Ketogenic amino acids Amino acids whose catabolism yields either acetoacetate or one of its precursor, (acetyl Co. A or acetoacetyl Co. A). Acetoacetate is one of the "ketone bodies. Leucine and lysine are the only exclusively ketogenic amino acids found in proteins. Their carbon skeletons are not substrates for gluconeogenesis and, therefore, cannot give rise to the net formation of glucose or glycogen in the liver, or glycogen in the muscle.

A mino acids that form oxaloacetate asparagine and aspartate form oxaloacetate. • Some rapidly dividing leukemic cells are unable to synthesize sufficient asparagine to support their growth. This makes asparagine an essential amino acid for these cells, which therefore require asparagine from the blood. Asparaginase can be administered to treat leukemic patients. • 1 Asparaginase lowers the level of asparagine in the plasma Aspartate loses its amino group by transamination to form oxaloacetate.

B- Amino acids that form α-ketoglutarate 1 -Glutamine is converted to glutamate and ammonia by the enzyme glutaminase. Glutamate is converted to α-keto- glutarate by transamination or through oxidative deamination by glutamate dehydrogenase.

B- Amino acids that form α-ketoglutarate 2 - Proline is oxidized to glutamate Glutamate is transaminated or oxidatively deaminated to form α-ketoglutarate.

B- Amino acids that form α-ketoglutarate 3 - Arginine and Ornithine : Arginine is cleaved by arginase to produce ornithine. Ornithine is subsequently converted to αketoglutarate. - Mutations in ornithineaminotransferase: elevate ornithine and cause gyrate atrophy of the retina. Treatment involves restricting dietary arginine. .

4 - Histidine is oxidatively deaminated by histidase to urocanic acid, which subsequently forms N-formiminoglutamate (FIGlu, Figure. 6). FIGlu donates its formimino group to tetrahydrofolate, leaving glutamate, which is degraded as described above. [Note: Individuals deficient in folic acid excrete increased amounts of FIGlu in the urine, particularly after ingestion of a large dose of histidine. The FIGlu excretion test has been used in diagnosing a deficiency of folic acid. ] Catabliosm of L -histidine to -ketoglutarate. (H 4 folate, tetrahydrofolate. ) Histidase is the probable site of the metabolic defect in histidinemia.

C. Amino acids that form pyruvate 1. Glycine can either be converted to serine by addition of a methylene group from N 5, N 10 methylenetetrahydrofolic or oxidized to CO 2 and NH 4+. 2. Serine Following conversion to glycine catalyzed by serine Hydroxyl methyltransferase [Note: The role of tetrahydrofolaten the transfer of one-carbon units Serine can also be converted to pyruvate by serine dehydratase

3. Alanine Transamination of -alanine forms pyruvate. Probably on account of its central role in metabolism there is no known metabolic defect of alanine catabolism.

4 - Cystine is reduced to cysteine by cystine reductase, using NADH + H+ as a reductant 10). Cysteine undergoes desulfuration to yield pyruvate(Figure 11). There are numerous abnormalities of cysteine metabolism. Cystine, lysine, arginine, and ornithine are excreted in cystine-lysinuria (cystinuria), a defect in renal reabsorption of these amino acids. Apart from cystine calculi, cystinuria is benign.
. Oxidation of acetaldehyde to")
5. Threonine aldolase cleaves threonine to acetaldehyde and glycine (pyruvate). Oxidation of acetaldehyde to acetate is followed by formation of acetyl-Co. A

D. Amino acids that form fumarate Phenylalanine and tyrosine: Hydroxylation of phenylalanine leads to the formation of tyrosine. This reaction, catalyzed by phenylalanine hydroxylase, is the first reaction in the catabolism of phenylalanine. Thus, the metabolism of phenylalanine and tyrosine merge, leading ultimately to the formation of fumarate and acetoacetate. Phenylalanine and tyrosine are, therefore, both glucogenic and ketogenic.
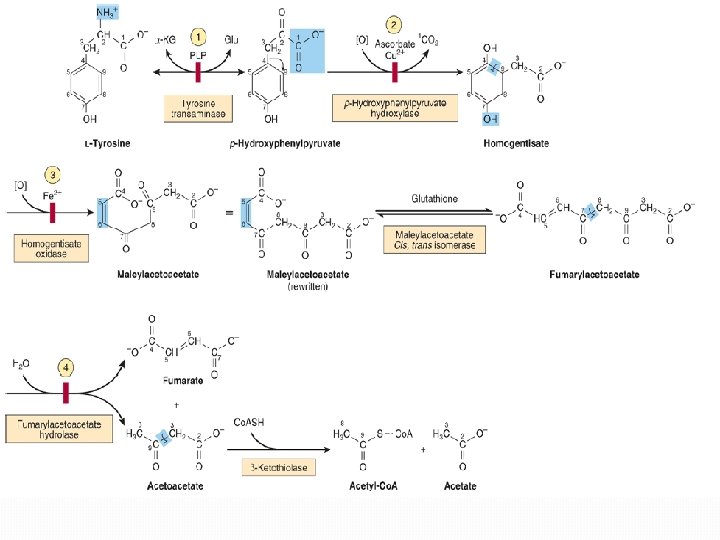

Inherited deficiencies in enzymes of phenylalanine and tyrosine metabolism lead to the diseases phenylketonuria : caused by a deficiency of phenylalanine hydroxylase Hyperphenylalaninemia : may be caused by deficiencies in the enzymes that synthesize or reduce the coenzyme tetrahydro- biopterin (BH 4). Alkaptonuria is a rare metabolic disease involving a deficiency in homogentisic acid oxidase, resulting in the accumulation of homogentisic acid [This reaction occurs in the degradative pathway of tyrosine]. Albinism refers to a group of conditions in which a defect in tyrosine metabolism results in a deficiency in the production of melanin.

E. Amino acids that form succinyl Co. A: 1. Methionine is one of four amino acids that form succinyl Co. A. This sulfur-containing amino acid deserves special attention because it is converted to Sadenosylmethionine (SAM). Methionine is also the source of homocysteine—a metabolite associated with atherosclerotic vascular disease. Formation of S -adenosylmethionine. CH 3 represents the high group transfer potential of "active methionine. "

1. Synthesis of SAM: Methionine condenses with ATP, forming SAM—a high-energy compound that is unusual in that it contains no phosphate. The formation of SAM is driven by hydrolysis of all three phosphate bonds in ATP (Figure 14). 2. Activated methyl group: The methyl group attached to the tertiary sulfur in SAMs "activated, " and can be transferred to a variety of acceptor molecules, such as ethanol amine in the synthesis of choline. The resulting loss of free energy accompanying the reaction makes methyl transfer essentially irreversible. 3. Hydrolysis of SAM: After donation of the methyl group, S-adenosylhomocysteine is hydrolyzed to homocysteine and adenosine. Homocysteine has two fates. If there is a deficiency of methionine, homocysteine may be remethylated to methionine. If methionine stores are adequate, homocysteine may enter the transsulfuration pathway, where is converted to cysteine. a. Resynthesis of methionine: Homocysteine accepts a methyl group from N 5 -methyl tetrahydrofolate (N 5 -methyl-THF) in a reaction requiring methylcobalamin, a coenzyme derived from vitamin B 12. The methy groups transferred from the B 12 derivative to homocysteine, and cobalamin recharged from N 5 -methyl-THF. b. Synthesis of cysteine: Homocysteine combines with serine, forming cystathionine, which is hydrolyzed to α-ketobutyrate and cysteine. This sequence has the net effect of converting serine to cysteine, and homocysteine toα-ketobutyrate, which is oxidatively decarboxylated to form propionyl Co. A. Propionyl Co. A is converted to succinyl Co. A. Because homocysteine is synthesized from the essential amino acid methionine, cysteine is not an essential amino acid as
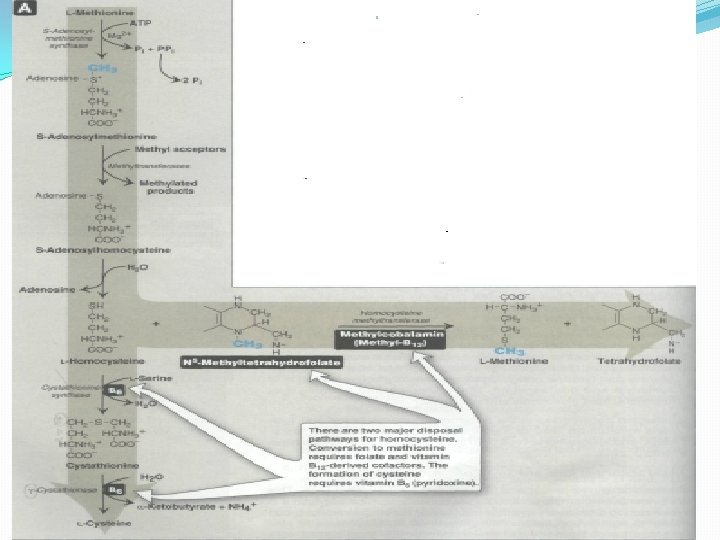

Role of homocysteine in vascular disease: Elevated plasma homocysteine levels are an independent cardiovascular risk factor that correlates with the severity of coronary artery disease. Dietary supplementation with folate, vitamin B 12 and vitamin B 6 the three vitamins involved in the metabolism of homocysteine —leads to a reduction in circulating levels of homocysteine. In patients with homocystinuria (characterized by high serum levels of homocysfeline caused by cystathionine synthase deficiency), experience premature vascular disease, and usually die of myocardial infarction, stroke, or pulmonary embolus. Thus, there is an association (but not a proven cause and effect relationship) of elevated homocysteine with cardiovascular disease.

E. Other amino acids that form succinyl Co. A Degradation of valine, isoleucine, and threonine also results in the production of succinyl Co. A a TCA cycle intermediate and glucogenic compound. 1. Valine and isoleucine are branched-chain amino acids that yield succinyl Co. A. 2. Threonine is dehydrated to α-ketobutyrate, which is converted to propionyl Co. A, the precursor of succinyl Co. A. [Note: Threonine can also be converted to pyruvate. ]

G. Amino acids that form acetyl Co. A or acetoacetyl Co. A Leucine, isoleucine, lysine, and tryptophan form acetyl Co. A or acetoacetyl Co. A directly. Phenylalanine and tyrosine also give rise to acetoacetate during their catabolism. Therefore, there a total of six ketogenic amino acids.

1. Leucine is exclusively ketogenic forming acetyl. Co. A and acetoacetate. 2. Isoleucine is both ketogenic and glucogenic, because its metabolism yields acetyl Co. A and propionyl Co. A. The first three steps in the metabolism of isoleucine are virtually identical to the initial steps in the degradation of the other branched-chain amino acids, valine and leucine 3. Lysine, an exclusively ketogenic amino acid, is unusual in that neither of its amino groups undergoes transamination as the first step in catabolism. Lysine is ultimately converted to acetoacetyl Co. A. 4. Tryptophan is both glucogenic and ketogenic because its metabolism yields alanine and acetoacetyl Co. A.

Catabolism of the branched-chain amino acids The branched-chain amino acids, isoleucine, and valine, are essential amino acids. These amino acids are metabolized primarily by the peripheral tissues (particularly muscle), rather than by the liver. Because these three amino acids have a similar route of catabolism, it is convenient to describe them as a group

1 -Transamination: Removal of the amino groups is catalyzed by branched-chain αamino acid aminotransferase. 2 - Oxidative decarboxylation: Removal of the carboxyl group of the αketo acids derived from leucine, valine, and isoleucine is catalyzed by a single enzyme complex, branchedchain α--keto acid dehydrogenase complex. An inherited deficiency of this enzyme results in accumulation of the branched-chain keto acid substrates in the urine. Their sweet odor prompted the name maple syrup urine disease.

Oxidation of the products formed in the above reaction yields (α-β unsaturated acyl Co. A derivatives. 4. End products: The catabolism of Isoleucine yields acetyl Co. A & succinyl Co. A (ketogenic & glucogenic). Valine yields succinyl Co. A (glucogenic). Leucine yields acetoacetate & acetyl Co. A (ketogenic). 3. Dehydrogenation:
- Slides: 26