Catabolism of Carbon Skeletons of AAs Prof Dr

Catabolism of Carbon Skeletons of AAs Prof. Dr. Arzu SEVEN

• The pathways of amino acid catabolism normally accounts for only 10 -15% of human body's energy production. • 20 catabolic pathways converge to form only 6 major products, all of which enter citric acid cycle. • From there, C skeletons are diverted to gluconeogenesis or ketogenesis or are completely oxidized to CO 2 and H 2 O.
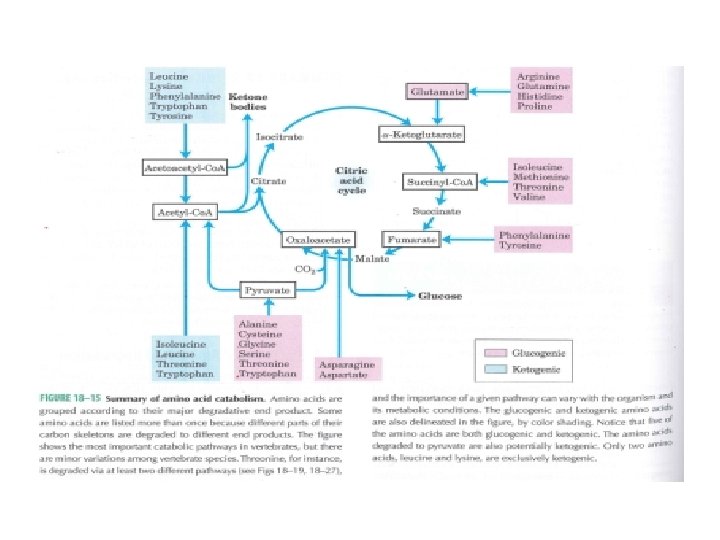

• Amino acids may be either glucogenic or ketogenic. • These amino acids that feed carbons into TCA cycle at the level of α-ketoglutarate, succinyl co. A, fumarate or oxaloacetate and those that produce pyruvate , can produce glucose via gluconeogenesis and are glucogenic (alanine, arginine, asparagine, aspartic acid, glycine, histidine, methionine, proline, serine, valine)

• Those amino acids that feed carbons at the level of acetyl-co. A or acetoacetyl co. A are ketogenic (leucine, lysine)

• Leucine is an exclusively ketogenic AA , its degradation makes a substantial contribution to ketosis under starvation • Both glucogenic and ketogenic AAs isoleucine, phenylalanine, threonine, tryptophan, tyrosine

• Amino acids that we can not synthesize are termed ESSENTİAL amino acids
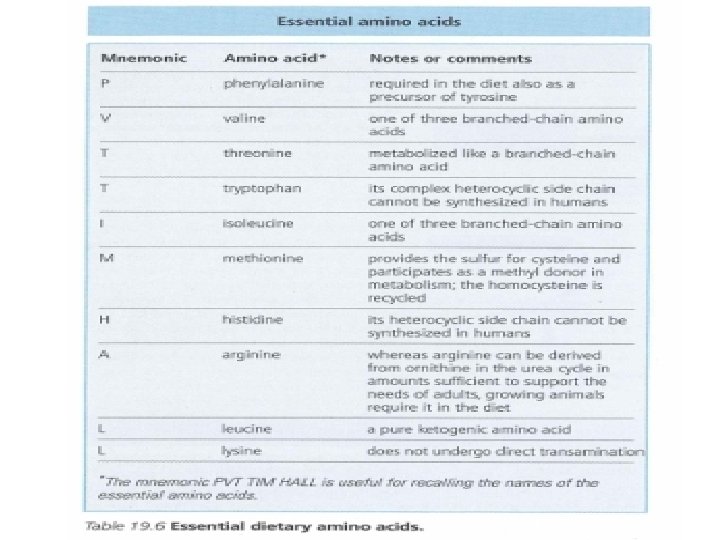

• Cysteine is not generally considered as an essential AA because it can be derived from non-essential amino acid serine, its sulfur must come from essential amino acid methionine. • Tyrosine is not required in the diet, but must be derived from essential amino acid phenylalanine.
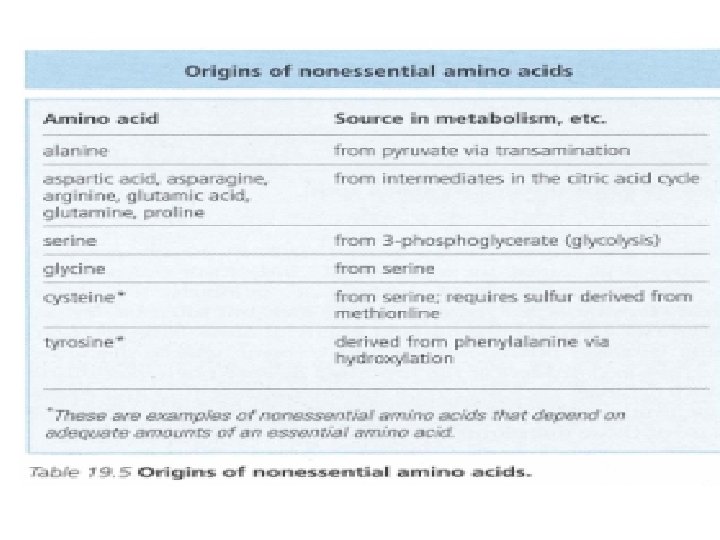

Conversion of AA to Specialized Products • Important products derived from AA include heme, purines, pyrimidines, hormones, neurotransmitters and biologically active peptides.

Glycine • Water-soluble glycine conjugates: glycocholic acid and hippuric acid formed from food additive benzoate. • Drugs or drug metabolites with carboxyl groups are excreted in the urine as glycine conjugates.

• Creatine and glutathione • Nitrogen and α-C of glycine are, incorporated into the pyrole rings and methylene bridge carbons of heme. • 4, 5, and 7 atoms of purine glycine is degraded via 3 pathways:
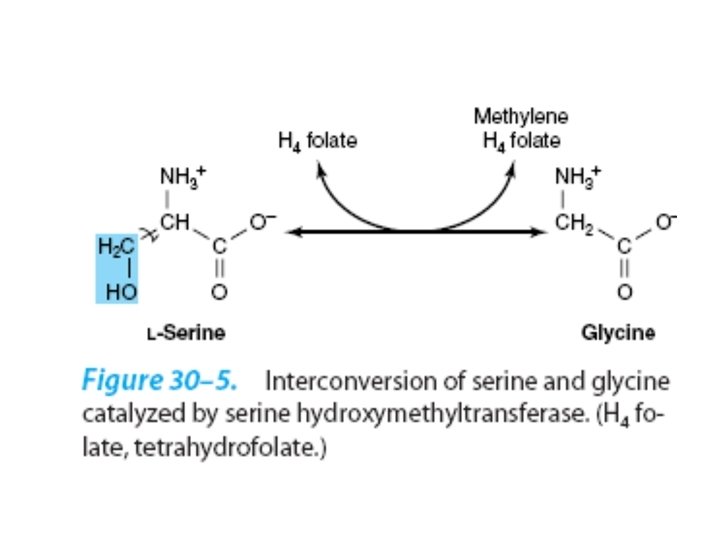
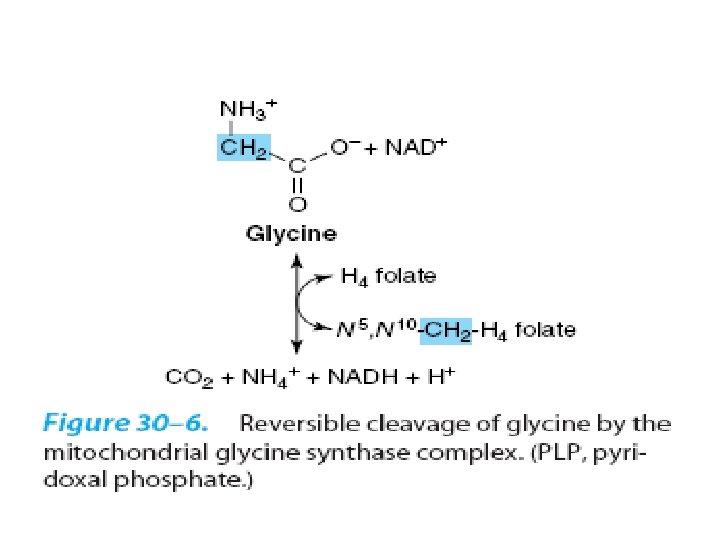
 •")
• Nonketotic hyperglycinemia: Defect in glycine cleavage enzyme activity • Glycine (serum) • mental deficiency • death in early childhood

• At high levels glycine is an inhibitory neurotransmitter. O H O NH • Glycine Glyoxylate NADHOxalate 2 2 D-Amino Acid oxidase 3

• Primary function of D-amino acid oxidase, present at high levels in the kidney, is to detoxify the ingested D-amino acids derived from bacterial cell walls and from grilled foodstuff. • Oxalate, from food or produced enzymatically in kidney, has medical significance as crystals of calcium oxalate in 75% of kidney stones. • (urolithiasis, nephrocalcinosis, early mortality from renal failure or hypertension)

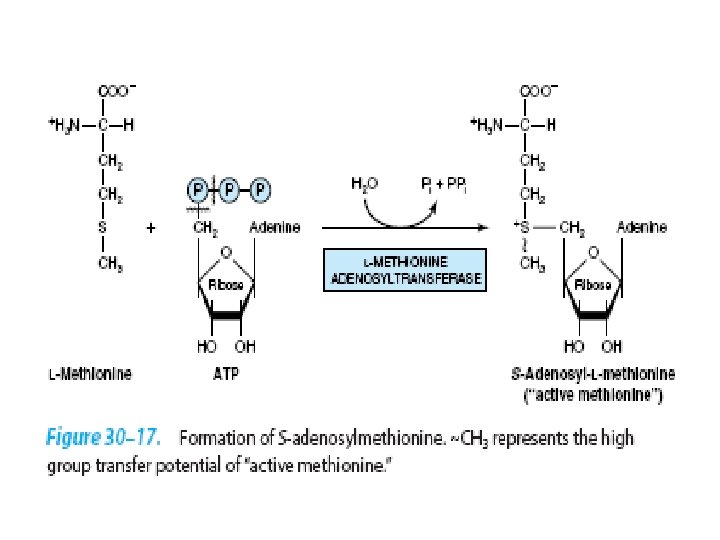
• Several enzyme cofactors play important roles in amino acid catabolism: Transamination requires pyridoxal phosphate • One Carbon transfer requires Biotin tetrahydrofolate and S-adenosylmethionine
 • Tetrahydrofolate transfers")
• Biotin transfers Carbon its most oxidized state (CO 2) • Tetrahydrofolate transfers one carbon groups in intermediate oxidation states (as methyl groups) • s-adenosylmethionine transfers methyl groups (the most reduced state of carbon)

Homocystinuria • A relatively rare autosomal recessive condition • Defect in methionine catabolism • Lack of an enzyme which catalyzes the transfer of sulfur from homocysteine to serine though the formation of cystathionine intermediate. • Mental retardation , vision problems, thrombotic strokes, coronary artery disease at young age.
 with")
• Defective carrier-mediated transport of cystine results in cystinosis (cystine storage disease) with deposition of cystine crystals in tissues and early mortality from acute renal failure. • In cystinuria, a defect in renal reabsorption, cystine, lysine, arginine and ornithine are excreted. • The mixed disulfide of L-cysteine and Lhomocysteine, excreted by cystinuric patients, is more soluble and reduces formation of cystine calculi.

• β-Alanine: • β-alanine, a metabolite of cysteine, is present in coenzyme A and as Balany. Ldipeptides (carnosine, anserine ) • Cysteine: • A precursor of thioethanol amine portion of coenzyme A • A precusor of taurine that conjugates with bile acids such as taurocholic acid
 Acid secretion İn stomach Histamine Allergic reaction vasodilatator")
Histidine • Histidin Decarboxylation (-co 2) Acid secretion İn stomach Histamine Allergic reaction vasodilatator

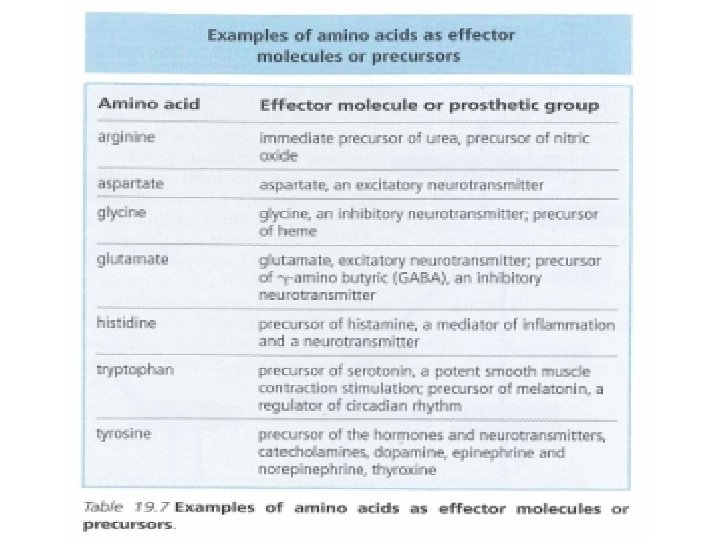
• Arginine: • Formamidine donor for creatine synthesis • Precursor of nitric oxide, NO (neurotransmitter, smooth muscle relaxant and vasodilatator)
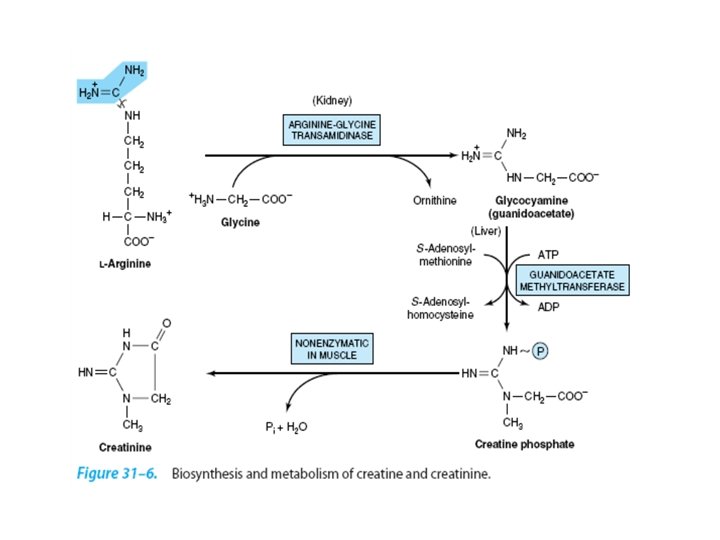

• Phosphocreatine, derived from creatine, is an important energy buffer in skeletal muscle. • Creatine is synthesized from glycine, arginine. • Methionine, in the form of S_adenosylmethionine, acts as a methyl donor.

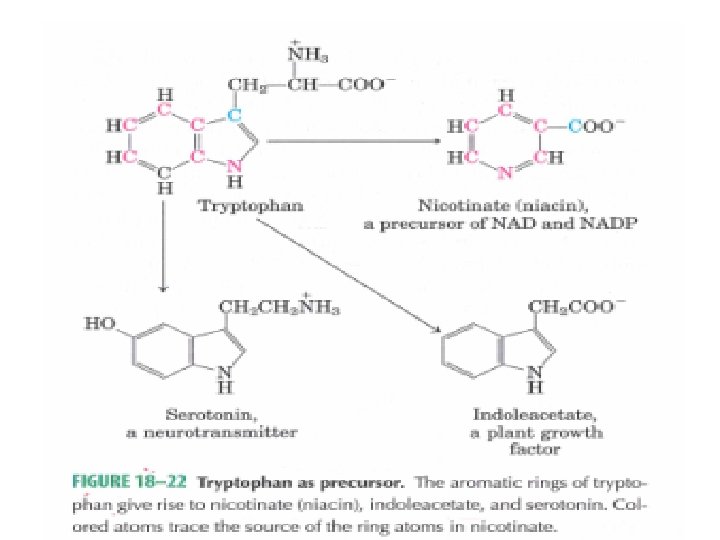
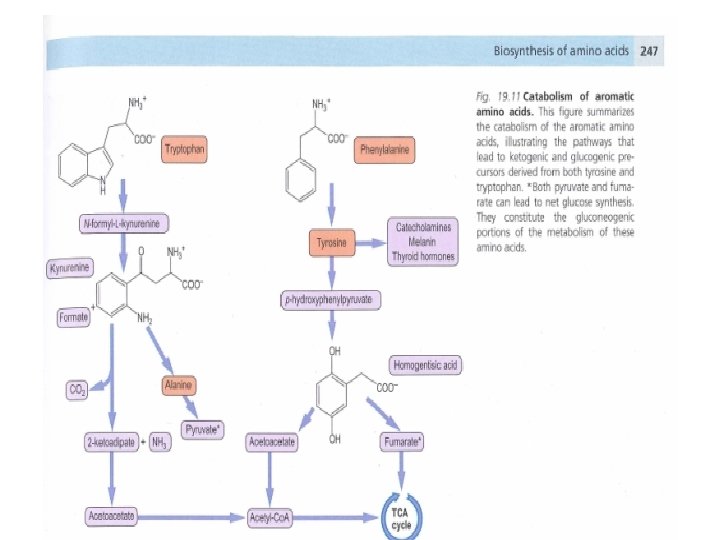
• Tryptophan, lysine, phenylalanine, tyrosine, leucine, isoleucine and threonine acetyl co. A and/or aceto acetyl -co. A • Tryptophan: Nicotinamide Serotonin indolacetate

• Principal normal urinary catabolites of tryptophan are 5 -hydroxyindolacetate and indole-3 -acetate.

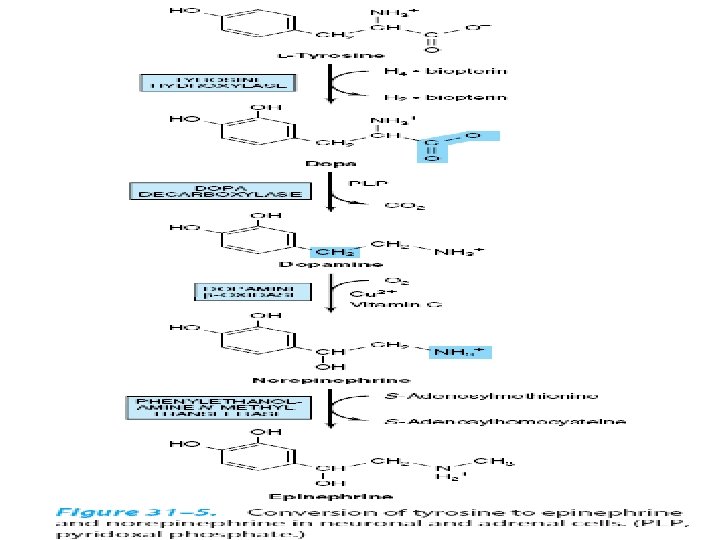
• Phenylalanine Tyrosine Dopamine NE E T 3, T 4 • Melanin is derived from tyrosine

• Parkinson's disease is associated with underproduction of dopamine. It has traditonally been treated by L-Dopa administration. • Over production of dopamine in the brain may be linked to schizophrenia.

• 5 hydroxytryptamine=serotonin: • A potent vasoconstrictor and stimulator of smooth muscle contraction.

• Serotonin N-acetylation O-methylation melatonin MAO Catalyzed oxidative deamination 5 hydroxy indolacetate
 • Tm cells that over produce serotonin.")
• Carcinoid(argentaffinoma) • Tm cells that over produce serotonin.

• Glutamate decarboxylation gives rise to GABA, an inhibitory neurotransmitter • Its overproduction is associated with epilectic seizures. • GABA analogs are used in the treatment of epilepsy and hypertension.
 • Functions in the brain as an inhibitory neurotransmitter by")
• γ-aminobutyrate (GABA) • Functions in the brain as an inhibitory neurotransmitter by altering transmembrane potential differences. • L-glutamate GABA decarboxylase
 are oxidized as fuels primarily in")
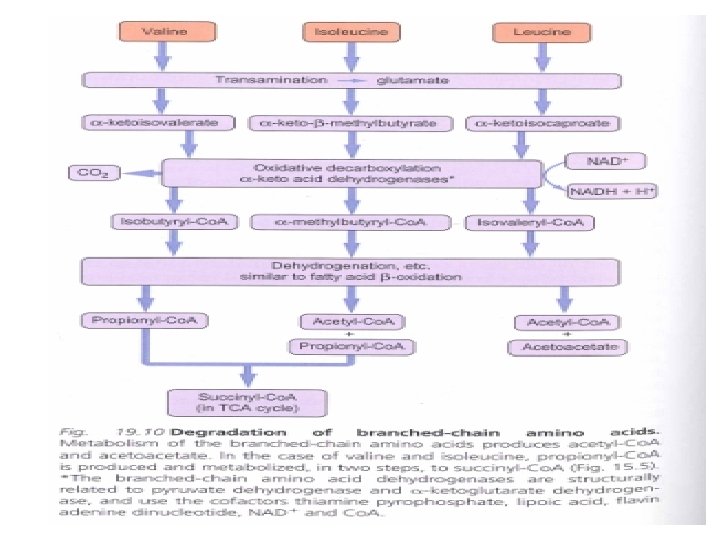
• Branched Chain AA (leucine, valine, isoleucine) are oxidized as fuels primarily in muscle, adipose, kidney and brain tissue (extrahepatic tissues)
 α- AA α- ketoacid")
• 1 -Transamınation (branced-chain amino transferase (absent in liver) α- AA α- ketoacid • 2 -Oxidative decarboxylation (by mitochondrial branched chain α-ketoacid dehydrogenase)

• This multimeric enzyme complex resembles pyruvate dehydrogenase and α -ketoglutarate dehydrogenase being inactivated by phosphorylation and activated by dephosphrylation. • 5 cofactors (TPP, FAD, NAD, lipoate, coenzyme A) • 3 -Dehydrogenation
- Slides: 45