Medical University of Sofia Faculty of Medicine Department





 diffusion The lipid soluble unionized drug diffuse acros the lipid biomembrane in")



 Facilitated diffusion. This proceeds more than passive (simple) diffusion and translocate even nondiffusible")




 - ileum (B 12, A,")
")

 and est have been use in this manner. Glucocorticost (GCS) applied")

 AUC – area under the curve AUC p. o. F –")




 It is accept that the body behaves as a")


 -redistribution in muscle and fat (long postnarcotic sleep)")
: includes the capil dothelial cells (which have tight junctions and")

 DDC МАО COMT 70%")

. Most drugs p physicochemical affinity for plasma proteins. Acid drugs")
 The drugs with high physicochemical affinity for plasma proteins (e. g. aspirin, sulfonamides,")


 Active metabolite from an active drug. Man are converted to one or more")
 and II (synthetic, conjuga")



 Hydrolysis. This is cleavage of drug molecule taking up of a molecule of")
 reacti These involve conjugation of the drug or its")


 Methylation. The amines and phenols can be methylated. Methionine and cysteine act as")
 METABOLIS This refers to metabolism of a drug during its pas")


 parallels")



")

")

 • morphine p. Kb: 7. 9 • stomach p. H:")
 of a drug is")
 kinetics. For majority o the processes involved in elimination are not")

 kinetics. In a few cases where the drugs are inactivat by")

 is the time in which the concentration of a")

- Slides: 70
Medical University of Sofia, Faculty of Medicine Department of Pharmacology and Toxicology GENERAL PHARMACOKINETICS Assoc. Prof. I. Lambev E-mail: itlambev@mail. bg
Pharmacokinetic – how the human body act on the drugs?
Pharmacokinetics is the quantitative stud drug movement in, through and out of the b Intensity of effect is related to concentratio the drug at the site of action, which depend on its pharmacokinetic properties. Pharmacokinetic properties of drug dete the route(s) of administration, dose, latency onset, time of peak action, duration of actio and frequency of drug administration. (KD Tripathy, 2007).
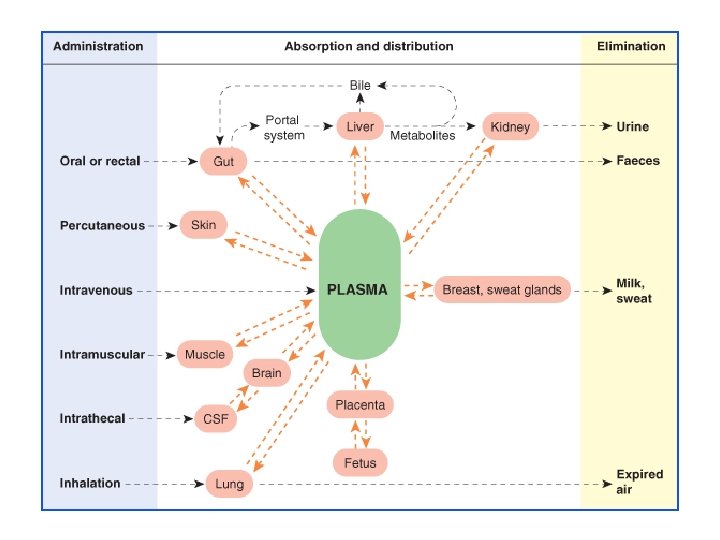
All pharmacokinetics processes involve transpor of the drug across biological lipid membrane.
Passive transport Passive diffusion Filtration Specialized transport Carrier transport Active transport Facilitated diffusion Pinocytosis etc.
Passive (simple) diffusion The lipid soluble unionized drug diffuse acros the lipid biomembrane in the direction of their concentration gradient. It does not need energ Most drugs are week electrolytes. Their ioniza is p. H dependent. The ionization of a week acid is given by the equation of Henderson–Hassel [AH] p. Ka = p. H + log 10 -----[A ]
p. Ka is the negative logarithm of acidic dissoc constant of the week electrolyte. If the conce of unionized drug [AH] is equal to concentratio ionized drug [A-], then [AH] ----- = 1 [A -] since log 1 is 0, under this condition p. H = p. Ka In this case the molecules of drugs are 50% io For a week base: p. Kb = p. H + [BH+] log 10 -----[B]
Filtration is passage of drug through aqueous in the membrane through paracelullar spaces. The moving force is hydrostatic or osmotic pr Lipid insoluble drugs cross biomembrane by filtration only if their molecular size is smaller the diameter of the enlarge aqueous pores. The filtration has an importance mainly at the of renal glomerul, where the size of capillaries large pores (40 Å) and most drugs (even album can filtrate. The brain capillary pores have sma
Carrier transport – by combination with a carrier molecule which a ferry-boat across the lipid region of the mem Carrier transport is saturable and competitivel bited by analogues which utilize the same carr a) Active transport is a movement against th b) centration gradient. It needs energy and is i c) by metabolic poisons. d) Levdopa and methyldopa are actively absor e) from the gut by aromatic amino acid transpo
b) Facilitated diffusion. This proceeds more than passive (simple) diffusion and translocate even nondiffusible substrates, but along their concentration gradient, therefore, does not need energy. Example: Facilitated transport of Pinocytosis involves the invagination of a of the cell membrane and trapping within the of a small vesicle containing extracellular con tuents. The vesicle contents can than be relea within the cell, or extruded from the other sid the cell. Pinocytosis is important for the trans of some macromolecules (e. g. insulin through
I. ABSORPTION It is the passage of drug from the s of administration into the circulatio Aqueous solubility. Drugs given in solid for dissolve in the aqueous biophase before they absorbed. For poorly water soluble drugs (asp griseofulvin) rate of dissolution governs rate o absorption. If a drug given as water solution, absorbed faster than when the same given in form or as a oily solution.
Concentration. Passive transport depends o concentration gradient. Drug given as concen solution is absorbed faster than from dilute s Area of absorbing surface. If the area is la the absorption is faster. Vascularity of absorbing surface. Blood c lation removes the drug from the site of abso and maintains concentration gradient across membrane. Increased blood flow hastens drug absorption.
Route of administration affects absorption, because each route h own peculiarities. Oral application. Unionized lipid soluble drugs ethanol) are readily absorbed from GIT. Acid drugs rin, barbiturates etc. ) are predominantly unionized in acid gastric juice and are absorbed from stomach. Acid drugs absorption from stomach is slower, beca mucosa is thick, covered with mucus and the surfac Basic drugs (e. g. atropine, morphine etc. ) are large zed and are absorbed only from the duodenum.
Presence of food dilutes the drug and retards ab Certain drugs form poorly absorbed complexes with constituents, e. g. tetracyclines with calcium presen Food delays gastric emptying. The most drugs are bed better if taken in empty stomach. Highly ionize e. g. amikacin, gentamicin, neostigmine, are poorly absorbed when given orally. Certain drugs are degraded in the GIT, e. g. penicil by acid, insulin by peptidases, and are ineffective o Enteric coated tablets (having acid resistant coatin sustained released preparations can be used to ov acid ability, gastric irritancy and brief duration of ac
Intestinal absorption: 2+ - duodenum (B 1, Fe ) - ileum (B 12, A, D, E, K) - large intestine + + (water, Na , Cl , K )
Drugs can also alter absorption by gut wall altering motility (atropine, amitriptyline, pethidine methoclopramide) or causing mucosal damage (neomycin, methotrexate, reserpine, vinblastine). Alteration of gut flora by antibiotics may disru enterohepatic recirculation of oral contraceptive and digoxin. S. c. and i. m. application By these routes the drugs is deposited in the vic the capillaries. Lipid soluble drugs pass readily a the whole surface of the capillary endothelium, bu very large molecules are absorbed through lymph
Many drugs not absorbed orally are absorbed pare Absorption from s. c. site is slower than that from i. but both are generally faster and more predictable p. o. absorption. Application of heat and muscular accelerate drug absorption by increasing blood flo Application of vasoconstrictors (e. g. adrenaline) re absorption. Many depot preparations (preparation long action), such as benzatine benzylpenicillin an protamine zinc insulin can be given by these route Topical applications (skin, cornea, mucous membranes) Systemic absorption depends on lipid solubility. Only a few drugs significantly penetrate intact skin.
Nitroglycerine, hyoscine (scopolamine) and est have been use in this manner. Glucocorticost (GCS) applied over extensive areas can produc systemic effects and pituitary-adrenal suppress Cornea is permeable to lipid soluble, unionized stigmine but not to highly ionized neostigmine. Similarly, mucous membrane of mouth, rectum vagina absorb lipophilic drugs, e. g. estrogen cr applied intravaginally has produced gynecomas in the male partner.
Bioavailability refers to the rate and extent of absorption of a drug from dosage form as determi by its concentration-time curve in blood or by its e in urine. It is a measure of the fraction (F) of admi dose of a drug that reaches the systemic circulatio unchanged form. Bioavailability of drug injected i. v. is 100%, bu frequently lower after oral ingestion, because: a) The drug may incompetely absorb b) The absorbed drug may undergo first pass c) metabolism in intestinal wall and/or liver d) excreted in bile.
Plasma concentration (mcg/ml) AUC – area under the curve AUC p. o. F – bioavailability F = ------ x 100% AUC i. v. (i. v. application) (p. o. application) 0 5 Time (h) 10 15
Plasma concentration time curves of the three preparations of a dr which containing the same amount. Formulation B is more slowly than A and may not produced therapeutic effect. Formulation C is to lesser extent (it has lower bioavailability).
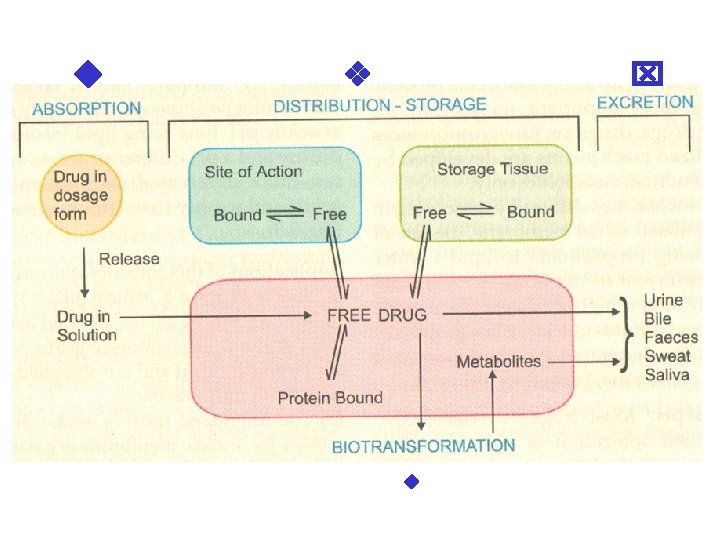
II. DISTRIBUTION It is the passage of drug from the circula to the tissue and site of its action. The extent of distribution of drug depends on its solubility, ionization at physiological p. H (depend p. K), extent of binding to plasma and tissue prote and differences in regional blood flow, disease li CHF, uremia, cirrhosis. Movement of drug proceeds until an equilibration established between unbound drug in plasma an tissue fluids.
Body fluid compartments The total body water as a percentage of bo mass varies from 50% to 70%, being rather less in women than in man. Body water is distributed into the follow main compartments: 1. plasma (5% of body mass) 2. intestinal fluid (16%) 3. intracellular fluid (35%) 4. transcellular fluid (2%) 5. fat (20%)
Two-compartment pharmacokinetic model
Apparent volume of distribution (Vd) It is accept that the body behaves as a singl homogeneous compartment with volume (V which drug gets immediately distributed: Dose administered i. v. Vd =Plasma --------------concentration
Drugs extensively bound to plasma proteins are restricted to the vascular compartment and have (e. g. warfarin – 99% bound and its Vd is 0, 1 L/k Drugs sequestrated in other tissues may have V more than total body water or even body mass, digoxin (6 L/kg) and propranolol (3 to 4 L/kg) be most of the drug is present in other tissues, and concentration is low. Therefore, in case of poisoning, drugs with la Vd are not easily removed by haemodia
Redistribution. Highly lipid soluble drugs give or by inhalationally get distributed to organs with blood flow (brain, heart, kidney, liver). Later they distributed to less vascular tissues (muscles and the drug-plasma concentrations falls. Greater the lipid solubility of the drug hastens its redistribution. Anaesthetic action of thiopentone (thiopental) is terminated in few minutes due to redistribution. However, when the same drug is repeatedly or continuously over long periods the perfusion high capacity sites get progressively fi up and the drug becomes longer acting.
Thiopental (thiopentone) -redistribution in muscle and fat (long postnarcotic sleep)
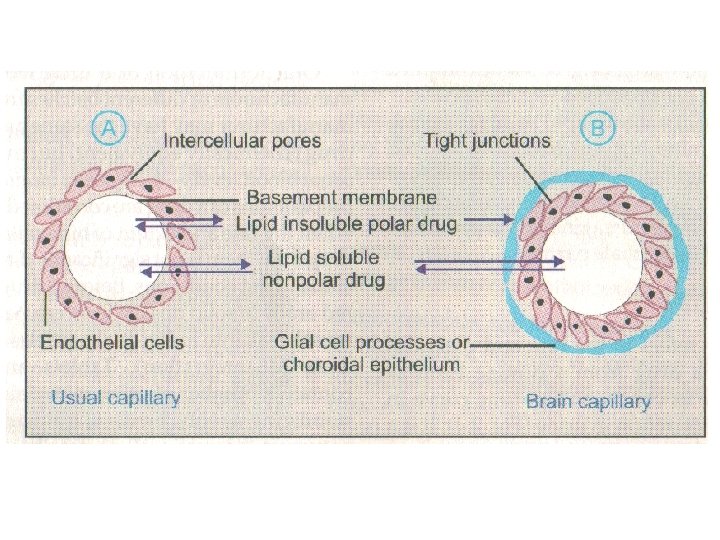
Blood brain barrier (BBB): includes the capil dothelial cells (which have tight junctions and lack intracellular pores) and an investment of glial tissu over the capillaries. A similar barrier is loctated in the choroid plexus.
BBB is lipoidal and limits the entry of non-lipid so drugs (amikacin, gentamicin, neostigmine etc. ). Only lipid soluble unionized drugs penetrate a have action on the CNS. Efflux carriers like P-gp (glycoprotein) present in capillary endothelial cells (also in intestinal muco renal tubular, hepatic canicular, placental and tes cells) extrude drugs that enter brain by other proc Inflammation of meanings of brain increases permeability of BBB. Dopamine (DA) does not enter brain, but its prec levodopa does. This is used latter in parkinsonism
GIT L-DOPA Blood and peripheral tissues Brain 1– 3% (Levodopa) DDC МАО COMT 70% 27– 29% C – DOPA-decarboxilase; COMT – catechol-О-methyltransfer
Placental barrier. Placental membranes are lip and allow free passage of lipophilic drug, while res hydrophilic drugs. The placental P-gp also serves limit foetal exposure to maternally administered dr However restricted amounts of nonlipid soluble dru when present in high concentration or for long per in maternal circulation, gain access to foetus. Thu is an incomplete barrier and many drug, taken by mother, can affect the foetus or the new born. Penicillins, azithromycin and erythromycin do not a the foetus and can use during the pregnancy.
Plasma protein binding (PPB). Most drugs p physicochemical affinity for plasma proteins. Acid drugs bind to plasma albumin and basic drugs to α 1 -glycoprotein. Extent of binding depends on dividual compound. Increasing concentration of dr progressively saturate the binding sites. The clini significant implications of PPB are: a) Highly PPB drugs are largely restricted to the v compartment and tend to have lower Vd. b) The PPB fraction is not available for action. c) There is an equilibration between PPB fraction and free molecules of drug.
d) The drugs with high physicochemical affinity for plasma proteins (e. g. aspirin, sulfonamides, chloramphenicol) can replace the other drugs (e. g. acenocoumarol, warfarin) or endogenous compounds (bilirubin) with lower affinity. e) High degree of protein binding makes the drug l acting, because bound fraction is not available f metabolism, unless it is actively excreted by live or kidney tubules. f) Generally expressed plasma concentrations of th refer to bound as well as free drug. g) In hypoalbuminemia, binding may be reduced a concentration of free drug may be attained (e. g.
Tissue storage. Drugs may also accumulate in organs or get bound to specific tissue constituents Heart and skeletal muscles – digoxin (to muscle Liver – chloroquine, tetracyclines, digoxin Kidney – digoxin, chloroquine Thyroid gland – iodine Brain – chlorpromazine, isoniazid, acetazolamide Retina – chloroquine (to nucleoproteins) Iris – ephedrine, atropine (to melanin) Bones and teeth – tetracyclines, heavy metals (to mucopolysaccharide of connective t Adipose tissues – thiopental, ether, minocycline,
III. METABOLISM (BIOTRANSFORMA Metabolism includes chemical alteration of the dru the body. Most hydrophilic drugs (amikacin, genta neostigmine, mannitol) are not biotransformated a excreted unchanged. Mechanism which metaboliz is developed to protect the body from toxins. The site for drug metabolism is liver, other sites are kid intestine, lungs and plasma. Metabolism of drugs may lead to the follo a) Inactivation. Most drugs and their active meta are converted to less active or inactive metabolite phenobarbital, morphine, propranolol etc.
b) Active metabolite from an active drug. Man are converted to one or more active metabolites diazepam, amitriptyline). c) Activation of inactive drug. Few drugs (so ca prodrugs) are inactive as such. They need conv in the body to one or more active metabolites (e. g vodopa, benfothiamine, enalapril, perindopril). The prodrug may offer advantages: their active may be more stable; they can have better bioava (e. g. benfothiamine), or other desirable pharmacokinetic properties or less side effects and toxicity.
Biotransformation reactions can be classified in phases: I (no synthetic) and II (synthetic, conjuga Phase I (no synthetic reactions) a) Oxidation is the most important drug metaboli reaction. Various oxidation reactions are hydroxy oxygenation at C-, N- or S-atoms; N or 0 -dealkyla oxidative deamination, etc. Oxidative reactions ar carry out by a group of monooxygenases in the liv which in the final step involve cytochrom P 450 reductase and O 2. There are more than 200 cytoc P 450 isoenzimes, differing in their affinity for vario substances (drugs). They are grouped into >20 fa
The majority of enzymes concerned with human metabolism belong to families CYP 1, 2 and 3. CYP 3 A 4/5 carry out biotransformation of largest number (30– 50%) of drugs. In addition to liver, this isoforms are expressed in intestine (responsible for first pas metabolism at this site) and kidney too. Inhibition CYP 3 A 4 by erythromycin, clarithromycin, ketocon itraconazole, verapamil, diltiazem and a constituen grape fruit juice is responsible for unwanted intera with terfenadine and astemizole. Rifampicin, phen carbmazepine, phenobarbital are inducers of the C
Barbiturates, phenothiazines, paracetamol, strero phenytoin, benzodiazepines, theophyllin and man drugs are oxydaized by CYP 450. Some other dru (adrenaline, mercaptopurine) and ethanol are oxid by mitochondrial or cytoplasmic enzimes. b) Reduction. This reaction is conversed of oxida involves CYP 450 enzymes working in the oppo direction. Drugs, primarily reduced, are chloram nicol, levodopa, halothane. DOPA-decarboxylase Levodopa (DOPA) Do
c) Hydrolysis. This is cleavage of drug molecule taking up of a molecule of water. Esterase Ester + H 20 Acid + Alcohol Similarly amides and polypeptides are hydrolyze by amidase and peptidases. Hydrolysis occurs i intestines, plasma and other tissues. Examples choline esters, procaine, lidocaine, pethidine, ox d) Cyclization is formation of ring structure fro straight chain compound, e. g. proguanil. e) Decyclization is opening up of ring structur the cyclic molecule, e. g. phenytoin, barbiturat
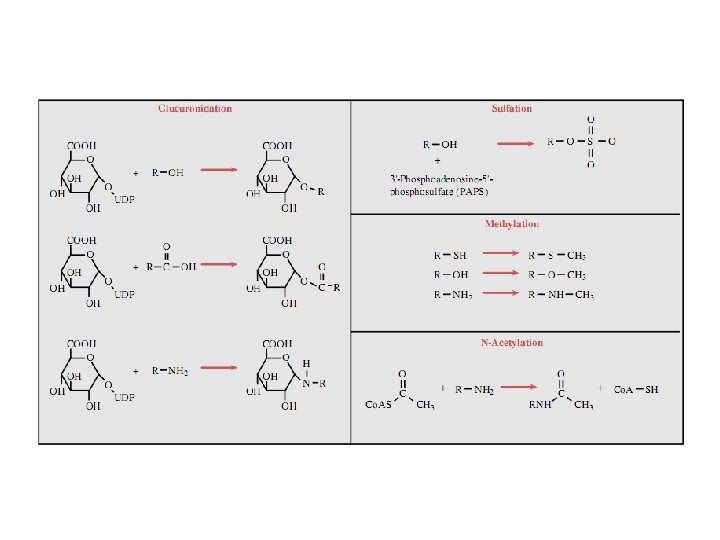
Phase II – synthetic (conjugation) reacti These involve conjugation of the drug or its phase bolite with an endogenous substrate to form a pola ionized organic acid, which is easily excreted in ur bile. Conjugation reactions have high energy requ (1) Glucoronide conjugation is the most importa thetic reaction. Compounds with a hydroxyl or carb acid group are easily conjugated with glucronic ac derived from glucose. Examples: chloramphenicol morphine, metroniazole, GCS, bilirubin, thyroxine. Drug glucuronides, excreted in bile, can be hydroly in the gut by bacteria, producing beta-glucoronidas
The liberated drug is reabsorbed and undergoes th fate. This enterohepatic recirculation of some dr chloramphenicol, phenolphthalein, oral contracept prolongs their action. (2) Acetylation. Compounds having amino or hyd residues are conjugated with the help of acetyl Co sulfonamides, isoniazid. Multiple genes control the transferases and rate of acetylation shows genetic morphizm (slow and fast acetylators). (3) Sulfate conjugation. The phenolic compounds steroids are sulfated by sulfokinases, e. g. chloram nicol, adrenal and sex steroids.
The two phase of drug metabolism
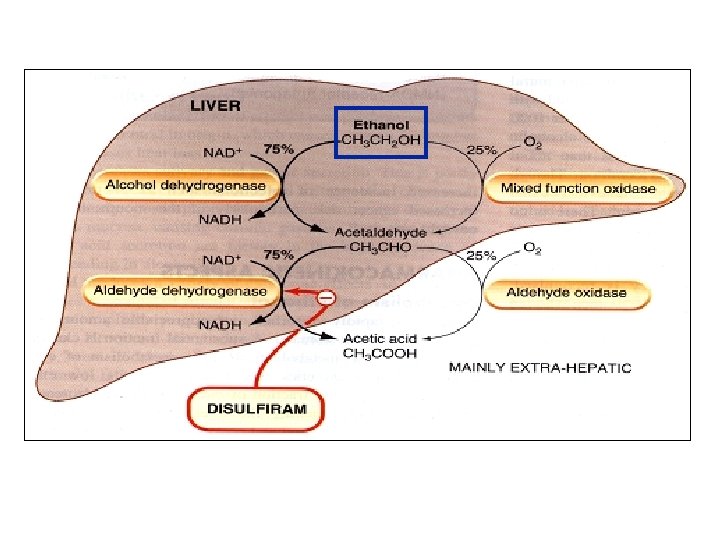
(4) Methylation. The amines and phenols can be methylated. Methionine and cysteine act as methy Examples: adrenaline, histamine, nicotinic acid. (5) Ribonucleoside/nucleotide synthesis is imp for the activation of many purine and pyrimidine an bolites used in cancer chemotherapy, e. g. Xeloda (6) Only a few drugs are metabolized by enzym intermediary metabolism. Examples: • alcohol by dehydrogenases • allopurinol by xanthine oxidase • succinylcholine and procaine by plasma cholines • adrenaline by monoamine oxidase (MAO)
FIRST PASS (PRESYSTEMIC) METABOLIS This refers to metabolism of a drug during its pas from the site of absorption into systemic circulatio orally administered drugs are exposed to drug me lism in the intestinal wall and liver in different exte • High first pass metabolism: propranolol, verapa pethidine, salbutamol, nitroglycerine, morphine, li • Orally dose of these drugs is higher than sublingu parenteral dose. • There is individual variation in the oral dose due to differences in the extent of first pass metabolism. • Oral bioavailability is increased in patients with se liver disease.
IV. EXCRETION Excretion is the passage out of systematically absorbed drug. Drug and their metabolites are excreted in: • urine (through the kidney) • bile and faeces • exhaled air • saliver and sweat • milk
The kidney is responsible for excreting of all water soluble substances. Glomerular filtration. Glomerul capillaries have lar All nonprotein bound drugs (lipid soluble or insoluble sented to the glomerulus are filtrated. Glomerular filt drugs depends on their plasma protein binding and r blood flow. Glomerular filtration rate (g. f. r. ) declines progressively after the age of 50 and is low in renal Tubular reabsorption. Lipid soluble drugs filtrated glomerulus back diffuse in the tubules because 99 glomerular filtrate is reabsorbed, but nonlipid solub and highly ionized drugs are unable to do so.
Thus, rate of excretion of such drugs, e. g. aminog (amikacin, gentamicin, tobramycin) parallels g. f. r. Changes in urinary p. H affect tubular reabsorption partially ionized drugs: • Weak bases ionize more and are less reabsorb in acidic urine. • Weak acids ionized more and are less reabsor in alkaline urine. This principle is utilized for facilitating elimina of drugs in poisoning: • Urine is acidified in morphine and atropine poison • Urine is alkalized in barbiturate and salicylate poi
The effect of changes in urinary p. H on drug excr is greatest for drug having p. Ka values between 5 because only in these case p. H dependent passiv reabsorption is significant. Tubular secretion is the active transfer of organ and bases by two separate nonspecific mechanis which operated in the proximal tububules: • Organic acid transport for penicillins, probenecid salicylates, uric acid, sulfinpyrazones, nitrofuran methotrexate, drug glucuronides etc. • Organic base transport for thiazides, quinine, pr amide, cimetidine, amiloride etc.
Many drug interactions occur due to competitio for tubular secretion, e. g. : • Aspirin blocks uricosuric action of probenecid and s pyrazone and decreases tubular secretion of metho • Probenecide decreases the urine concentration of n toin, increases the duration of penicillin action and secretion of methotrexate. • Quinidine decreases renal and biliary clearance of by inhibiting efflux carrier P-gp. Tubular transport mechanisms are no well develope Duration of action of many drugs (penicillins, cephal ins, aspirin etc. ) is longer in neonates. These system mature during infancy.
• aminoglycosides • beta-lactams • sulfonamides • quinolones • nitrofurans • polymyxins
• macrolides • lincosamines • rifampicin • tetracyclines (p. o. )
• General inhalation anaesthetics • Potassium iodide • Bronchoantiseptic oils • Alcohol
• sulfonamides • barbiturates • reserpine • alcohol • Coffeinum (Caffeine)
Saliva excretion • oleandomycin • spiramycin • phenytoin • zalcitabine • verapamil
Morphine (10% stomach excretion) • morphine p. Kb: 7. 9 • stomach p. H: 1– 2 • plasma p. H: 7. 36
KINETICS OF ELIMINATION (elimination = metabolism + excreti Clearance (Cl) of a drug is theoretical volume from which the drug is completely removed in unit Cl = Rate of elimination/Plasma concentratio Renal (Clr) or creatinine clearance (Clcr): Curine x Vurine Clrenal = ----------Cplasma
First order (exponential) kinetics. For majority o the processes involved in elimination are not satur over the clinically obtained concentrations. These have first order kinetics. Their rate of elimination is directly proportional to drug concentration and the rance remains constant.
Semilog plasma concentration-time plot of a drug eliminated by first order kinetics after i. v. injection.
Zero order (linear) kinetics. In a few cases where the drugs are inactivat by metabolic degradation (such as ethanol, phenytoin, theophylline, salicylates and warf the time-course of disappearance of drug fro the plasma does not follow the exponential o biexponential pattern, but is initially linear. These drugs are removed at a constant rate which is independent of plasma concentratio This is often called zero order kinetics.
The blood alcohol concentration falls linearly and the rate of fall does not vary with dose.
Plasma half live (t 1/2) is the time in which the concentration of a drug declines by one half. Drug long t 1/2 can accumulate. Plasma t 1/2 of some drug Adenosine < 2 sec Dobutamine – 2 min Benzylpenicillin – 30 min Amoxicillin – 1 h Paracetamol – 2 h Atenolol – 7 h Diazepam – 40 h Ethosuccimide – 54 h Digitoxin – 168 h
From the peak plasma concentration the drug is tually eliminated from the plasma in 5 t 1/2 period (1) (2) (3) (4) (5)