Introduction to Biostatistics and Bioinformatics http fenyolab orgibb

Introduction to Biostatistics and Bioinformatics http: //fenyolab. org/ibb 2015/ ibb 2015@fenyolab. org

Introduction to Biostatistics and Bioinformatics Lectures: Tuesdays and Thursdays 2 -4 pm in different locations: Skirball 3 rd and 4 th Floor Seminar Rooms and Smilow 1 st Floor Seminar Room. Check web site: http: //fenyolab. org/ibb 2015. Tutorials: Thursdays 4 pm in Room 718 in the Translational Research Building (227 E 30 th St between 2 nd and 3 rd). Homework: Given on Tuesdays and due on Sunday. Course Assessment Participation (20%) Assignments (40%) Exam (40%)

Introduction to Biostatistics and Bioinformatics Learning Objective • Good experimental design. • Automatically process data generated in the lab and combine it with public data. • Statistical methods for data analysis.

Introduction to Biostatistics and Bioinformatics Lectures • • • • Introduction to Biological Data Introduction to Python III Introduction to Python IV Exploring Data & Descriptive Statistics Sequence Alignment Concepts Sequence Database Searching Probability Distributions Estimation Hypothesis Testing Analysis of Variance Regression & Correlation Experimental Design & Analysis

This Lecture IBB_2015 Data types and representations in Molecular Biology

Learning Objectives • text formats for some common genomics data types • formatting text with tag: value pairs • basic database concepts • details of the FASTA format • Data formats in public molecular biology databases • Genbank, db. SNP • Genome Browsers: BED format • Database queries: field specific queries

Biologists Collect Lots of Data • Hundreds of thousands of species • Millions of articles in scientific journals • Genetic information: – gene names – phenotype of mutants – location of genes/mutations on chromosmes – linkage (distances between genes)

• High Throughput lab technology – PCR – Gene expression microarrays – Rapid inexpensive DNA sequencing – Many methods of collecting genotype data • Assays for specific polymorphisms • Genome-wide SNP chips • Must have data quality assessment prior to analysis

Data files • Various assay technologies/machines collect raw data in custom formats • Images • Trace files • Machine specific binary formats • Convert to text to share scientific data – Why text? • Does not require custom software to read the data • Stable for long periods of time across different computing systems (ASCII is universal) • Can be smoothly shared across many different computing systems – The WWW is built with text (html)

Text has many different formats FASTA >URO 1 uro 1. seq Length: 2018 November 9, 2000 11: 50 Type: N Check: 3854. . CGCAGAAAGAGGAGGCGCTTGCCTTCAGCTTGTGGG AAATCCCGAAGATGGCCAAAGACAACTGTTCG TTGCTTCCAGGGCCTGCTGATTTTTGGAAATGTGATT ATTGGTTGTTGCGGCATTGCCCTGACTGCGGAGTGC ATCTTCTTTGTATCTGACCAACACAGCCTCTACCCACT GCTTGAAGCCACCGACAACGATGACATCTATGGGGC TGCCTGGATCGGCATATTTGTGGGCATCTGCCTCTTC TGCCTGTTCTAGGCATTGTAGGCATCATGAAGT CCAGCAGGAAAATTCTTCTGGCGTATTTCATTCTGAT GTTTATAGTATATGCCTTTGAAGTGGCATCTTGTATCA CAGCAGCAACAAGACTTTTTCACACCCAACCT CTTCCTGAAGCAGATGCTAGAGAGGTACCAAAACAAC AGCCCTCCAAACAATGATGACCAGTGGAAAAACAATG FASTQ GFF 3 ##gff-version 3 #!gff-spec-version 1. 20 ##species_http: //www. ncbi. nlm. nih. gov/Taxonomy/Browser/ww wtax. cgi? id=7425 NC_015867. 2 Ref. Seq c. DNA_match 66086 66146. -. ID=aln 0; Target=XM_008204328. 1 1 61 +; for_remapping=2; gap_count=1; num_ident=8766; num_mismatc h=0; pct_coverage=100; pct_coverage_hiqual=100; pct_identity_ gap=99. 9886; pct_identity_ungap=100; rank=1 NC_015867. 2 Ref. Seq c. DNA_match 65959 66007. -. ID=aln 0; Target=XM_008204328. 1 62 110 +; for_remapping=2; gap_count=1; num_ident=8766; num_misma tch=0; pct_coverage=100; pct_coverage_hiqual=100; pct_identity _gap=99. 9886; pct_identity_ungap=100; rank=1 NC_015867. 2 Ref. Seq c. DNA_match 65799 65825. -. ID=aln 0; Target=XM_008204328. 1 111 137 +; for_remapping=2; gap_count=1; num_ident=8766; num_misma tch=0; pct_coverage=100; pct_coverage_hiqual=100; pct_identity _gap=99. 9886; pct_identity_ungap=100; rank=1 @SRR 350953. 5 MENDEL_0047_FC 62 MN 8 AAXX: 1: 1: 1646: 938 length=152 NTCTTTTTCCTCTTTTGCCAACTTCAGCTAAATAGGAGCTACACTGATTAGGCAGAAACTTGATTAACAGGGCTTAAGGTAACC TTGTTGTAGGCCGTTTTGTAGCACTCAAAGCAATTGGTACCTCAACTGCAAAAGTCCTTGGCCC +SRR 350953. 5 MENDEL_0047_FC 62 MN 8 AAXX: 1: 1: 1646: 938 length=152 +50000222 C@@@@@22: : 8888898989: : : <<<: <<<<: : : <<<<<: <: <<<IIIIIGFEEGGGGGGGII@IGDGBGGGGGGDDIIGIIE GIGG>GGGGGGDGGGGGIIHIIBIIIGIIIHIIIIGII @SRR 350953. 7 MENDEL_0047_FC 62 MN 8 AAXX: 1: 1: 1724: 932 length=152 NTGTGATAGGCTTTGTCCATTCTGGAAACTCAATATTACTTGCGAGTCCTCAAAGGTAATTTTTGCTATTGCCAATATTCCTCAGAGG AAAAAAGATACAATACTATGTTTTATCTAAATTAGCATTAGAAAATCTTTCATTAGGTGT +SRR 350953. 7 MENDEL_0047_FC 62 MN 8 AAXX: 1: 1: 1724: 932 length=152 #. , ')2/@@@@@<: <<: 778789979888889: : : 99999<<: : : : : <<<<<@@@@@: : : IHIGIGGGGGGDGGDGGDDDIHIHIIIII 8 GGG GGIIHHIIIGIIGIBIGIIIIEIHGGFIHHIIIIIIIGIIFIG

tag: value pairs • A very common way to organize text is with tag: value pairs • address: Publisher's address (usually just the city, but can be the full address for lesser-known publishers) • annote: An annotation for annotated bibliography styles (not typical) • author: The name(s) of the author(s) (in the case of more than one author, separated by and) • booktitle: The title of the book, if only part of it is being cited • chapter: The chapter number • HTML is a tag system to display text in web browsers. <b>This text is bold</b> <a href="http: //www. w 3 schools. com">This is a link</a> <h 1 style="font-family: verdana">This is a heading</h 1> <p style="color: green; margin-left: 20 px; ">This is a paragraph. </p>

What is a Database? • Structured data • Information is stored in "records" and "fields" • Fields are categories – Must contain data of the same type • Records contain data that is related to one object across fields • A record does not need to have data in every field • A record is a series of tag-value pairs where fields are the tags Unique Identifier SNP ID SNPSeq. ID Gene +primer -primer Hap A Hap B Hap. C D 1 Mit 160_1 10. MMHAP 6 7 FLD 1. seq lymphocyte antigen 84 AAGGTAAAA GGCAATCAG CACAGCC TCAACCTGG AGTCAGAG GCT C — A M-05554_1 12. MMHAP 3 1 FLD 3. seq procollagen, type III, alpha TGCGCAGAA GCTGAAGTC TA TTTTGAGGT GTTAATGGT TCT C — A M-05554_2 X 60184 complement component factor i ACTTCCAGC CCTGGCTCT ATATGCCAC CAAGAAGCA A C — M-09947_3 AF 067835 caspase 8 TCACAGAGG GAAACATGA AG CTCCACATT GAACCAAAG CA G C T M-11415_1 U 02023 insulin-like growth factor binding protein GGGAAAAG CCTGAAAGA AGCTGAAAC CGGACATCA AT T G — D 1 Mit 284_ 3 J 05234 nucleolin TGTTGGAAC CGACTTCTT CA AAGAGTCAA AGAATTTAT GGAATGA G T T

A Spreadsheet can be a Database • columns are Fields SNP ID • Rows are Records • Can search for a term within just one field • Or combine searches across several fields SNPSeq ID Gene +primer -primer Hap A Hap B Hap C D 1 Mit 160_1 10. MMHAP 6 7 FLD 1. seq lymphocyte antigen 84 AAGGTAAAA GGCAATCAG CACAGCC TCAACCTGG AGTCAGAGG CT C — A M-05554_1 12. MMHAP 3 1 FLD 3. seq procollagen, type III, alpha TGCGCAGAA GCTGAAGTC TA TTTTGAGGT GTTAATGGTT CT C — A M-05554_2 X 60184 complement component factor i ACTTCCAGC CCTGGCTCT ATATGCCAC CAAGAAGCA A C — M-09947_3 AF 067835 caspase 8 TCACAGAGG GAAACATGA AG CTCCACATT GAACCAAAG CA G C T M-11415_1 U 02023 insulin-like growth factor binding protein GGGAAAAGC CTGAAAGAA GC AGCTGAAAC CGGACATCA AT T G — D 1 Mit 284_ 3 J 05234 nucleolin TGTTGGAAC CGACTTCTTC A AAGAGTCAA AGAATTTATG GAATGA G T T

Spreadsheet data can be saved as tab or comma separated values Tab delimited csv Ovary embryo (0 -3 hrs)embryo (3 -6 hrs)embryo (6 -9 hrs)Pupal 0. 130666 0. 443061 0. 273747 0. 0643887 8. 93599 0. 229979 0. 0277432 1. 46693 0. 408315 0. 108006 0. 151759 0. 0793942 0. 139144 0. 0178557 0. 0144366 0. 0752579 0. 208803 0. 208607 0. 0298946 0. 0220337 0. 0361666 0. 0392186 0. 0572734 3. 82547 0. 129111 0. 180263 0. 476863 0. 412562 1. 44697 39. 1049 5. 28872 0. 219502 0. 738405 3. 4654 2. 53851 2. 08545 485. 993 5. 38445 1. 06842 6. 40536 3. 87577 22. 5721 58. 8561 77. 217 2. 07522 1. 34702 2. 5313 4. 1273 10. 0762 530. 451 27. 6188 35. 8966 7. 35556 4. 73383 16. 4197 0. 798709 0. 692811 10. 2703 6. 14537 3. 20737 3. 655 3. 29876 1. 24837 0. 23297 3. 07092 gene, Ovary, embryo(0 -3 hrs), embryo(3 -6 hrs), embryo(6 -9 hrs), Pupal LOC 100118025, 0. 04541333, 0. 006205798, 0. 165735055, 2. 226200589, 2. 556445228 LOC 100122637, 0. 233690353, 0. 007614514, 0. 217603805, 2. 044255893, 2. 496835435 LOC 100116733, 0. 033557481, 0. 009225546, 0. 177377903, 2. 76701782, 2. 012821249 LOC 100120954, 0. 003250874, 0. 010542103, 1. 974338817, 2. 971542769, 0. 040325437 LOC 100122540, 0. 483847049, 0. 01129521, 0. 286362403, 4. 180982477, 0. 037512862 LOC 100119626, 0. 089661159, 0. 01165491, 0. 085576525, 0. 809059218, 4. 004048189 Scr, 0. 016751983, 0. 013304455, 0. 445865943, 0. 813361695, 3. 710715923 LOC 100119924, 0. 685022497, 0. 016888969, 1. 618261922, 1. 182058753, 1. 49776786 LOC 100121348, 0. 18959044, 0. 018210136, 1. 178691029, 1. 916404302, 1. 697104093

Data Formats • How to organize various types of genetic data? • Need standard formats • DNA sequence = GATC, but what about gaps, unknown letters, etc. – How many letters per line – ? ? Spaces, numbers, headers, etc. – Store as a string, code as binary numbers, etc. • Use a completely different format for proteins?
,")
FASTA Format • In the process of writing a similarity searching program (in 1985), William Pearson designed a simple text format for DNA and protein sequences • The FASTA format is now universal for all databases and software that handles DNA and protein sequences One header line, starts with > with a [return] at end All other characters are part of sequence. Most software ignores spaces, carriage returns. Some ignores numbers >URO 1 uro 1. seq Length: 2018 November 9, 2000 11: 50 Type: N Check: 3854. . CGCAGAAAGAGGAGGCGCTTGCCTTCAGCTTGTGGGAAATCCCGAAGATGGCCAAAGAC A ACTCAACTGTTCGTTGCTTCCAGGGCCTGCTGATTTTTGGAAATGTGATTATTGGTTGTT GCGGCATTGCCCTGACTGCGGAGTGCATCTTCTTTGTATCTGACCAACACAGCCTCTACC CACTGCTTGAAGCCACCGACAACGATGACATCTATGGGGCTGCCTGGATCGGCATATTTG TGGGCATCTGCCTCTTCTGCCTGTTCTAGGCATTGTAGGCATCATGAAGTCCAGCA GGAAAATTCTTCTGGCGTATTTCATTCTGATGTTTATAGTATATGCCTTTGAAGTGGCAT CTTGTATCACAGCAGCAACAAGACTTTTTCACACCCAACCTCTTCCTGAAGCAGA TGCTAGAGAGGTACCAAAACAACAGCCCTCCAAACAATGATGACCAGTGGAAAAACAATG
; ID=FBpp")
Multi-Sequence FASTA file >FBpp 0074027 type=protein; loc=X: complement(16159413. . 16159860, 16160061. . 16160497); ID=FBpp 0074027; name=CG 12507 -PA; parent=FBgn 0030729, FBtr 0074248; dbxref=Fly. Base: FBpp 0074027, Fly. Base_Annotation_IDs: CG 12507 PA, GB_protein: AAF 48569. 1, GB_protein: AAF 48569; MD 5=123 b 97 d 79 d 04 a 06 c 66 e 12 fa 665 e 6 d 801; release=r 5. 1; species=Dmel; length=294; MRCLMPLLLANCIAANPSFEDPDRSLDMEAKDSSVVDTMGMGMGVLDPTQPKQMNYQKPPLGYKDYDYYLGSRRMADPYGADNDLSASSAIKIHGE GNLASLNRPVSGVAHKPLPWYGDYSGKLLASAPPMYPSRSYDPYIRRYDRYDEQYHRNYPQYFEDMYMHRQRFDPYDSYSPRIPQYPEPYVM YPDRYPDAPPLRDYPKLRRGYIGEPMAPIDSYSSSKYVSSKQSDLSFPVRNERIVYYAHLPEIVRTPYDSGSPEDRNSAPYKLNKKKIKNIQRPLA NNSTTYKMTL >FBpp 0082232 type=protein; loc=3 R: complement(9207109. . 9207225, 9207285. . 9207431); ID=FBpp 0082232; name=m. Rp. S 21 -PA; parent=FBgn 0044511, FBtr 0082764; dbxref=Fly. Base: FBpp 0082232, Fly. Base_Annotation_IDs: CG 32854 PA, GB_protein: AAN 13563. 1, GB_protein: AAN 13563; MD 5=dcf 91821 f 75 ffab 320491 d 124 a 0 d 816 c; release=r 5. 1; species=Dmel; length=87; MRHVQFLARTVLVQNNNVEEACRLLNRVLGKEELLDQFRRTRFYEKPYQV RRRINFEKCKAIYNEDMNRKIQFVLRKNRAEPFPGCS >FBpp 0091159 type=protein; loc=2 R: complement(2511337. . 2511531, 2511594. . 2511767, 2511824. . 2511979, 2512032. . 2512082); ID=FBpp 0091159; name=CG 33919 -PA; parent=FBgn 0053919, FBtr 0091923; dbxref=Fly. Base: FBpp 0091159, Fly. Base_Annotation_IDs: CG 33919 PA, GB_protein: AAZ 52801. 1, GB_protein: AAZ 52801; MD 5=c 91 d 880 b 654 cd 612 d 7292676 f 95038 c 5; release=r 5. 1; species=Dmel; length=191; MKLVLVVLLGCCFIGQLTNTQLVYKLKKIECLVNRTRVSNVSCHVKAINWNLAVVNMDCFMIVPLHNPIIRMQVFTKDYSNQYKPFLVDVKIRICEVIER RNFIPYGVIMWKLFKRYTNVNHSCPFSGHLIARDGFLDTSLLPPFPQGFYQVSLVVTDTNSTSTDYVGTMKFFLQAMEHIKSKKTHNLVHN >FBpp 0070770 type=protein; loc=X: join(5584802. . 5585021, 5585925. . 5586137, 5586198. . 5586342, 5586410. . 5586605); ID=FBpp 0070770; name=cv. PA; parent=FBgn 0000394, FBtr 0070804; dbxref=Fly. Base: FBpp 0070770, Fly. Base_Annotation_IDs: CG 12410 PA, GB_protein: AAF 46063. 1, GB_protein: AAF 46063; MD 5=0626 ee 34 a 518 f 248 bbdda 11 a 211 f 9 b 14; release=r 5. 1; species=Dmel; length=257; MEIWRSLTVGTIVLLAIVCFYGTVESCNEVVCASIVSKCMLTQSCKCELKNCSCCKECLKCLGKNYEECCSCVELCPKPNDTRNSLSKKSHVEDFDGVP ELFNAVATPDEGDSFGYNWNVFTFQVDFDKYLKGPKLEKDGHYFLRTNDKNLDEAIQERDNIVTVNCTVIYLDQCVSWNKCRTSCQTTGASSTR WFHDGCCECVGSTCINYGVNESRCRKCPESKGELGDELDDPMEEEMQDFGESMGPFD How many fields? What is the key, what is the value? Does the header have tag: value encoded data?

Other Standards? • Other types of important medical and genetic data may or may not have universal standards: • Genotype/haplotype • Clinical records • Gene expression • Genome annotation • Protein structure • Alignments • Phylogenetic trees

Where/How are Data Formats Defined? • Unfortunately, there is no single repository of Standards for important widely used bioinformatics data formats. • Each file type has its own peculiar history, and may or may not have a home database, or an official group that maintains and/or enforces a standard. • Gen. Bank format is defined by the NCBI Gen. Bank database. http: //www. ncbi. nlm. nih. gov/Sitemap/samplerecord. html • The BED format (for genome intervals) is defined by the UCSC Genome Browser: https: //genome. ucsc. edu/FAQformat. html#format 1 • The GFF 3 format is defined on the Sequence Ontology website: http: //www. sequenceontology. org/resources/gff 3. html • FASTA and FASTQ formats are “de facto” standards that are not formally defined or enforced by anyone: Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010 Apr; 38(6): 1767 -71. • I typically Google search “xyz file format definition”

Software File Formats • Within a single software language (eg. Python, Perl, Java, etc. ), file formats are rigorously defined as data types. • Thus we can know with certainty where in the file to find numbers, text, gene IDs, chromosome locations, etc. when we are writing a program. • There may be challenges when reading data into software from public sources that do not obey the same rigorous standard.

SNPStats Hap. Stat
 work of bioinformatics involves")
Reformatting Data Files • Much of the routine (yet annoying) work of bioinformatics involves messing around with data files to get them into formats that will work with various software • Then messing around with the results produced by that software to create a useful summary…

Public Databases • In addition to your own experimental data, access to public data is essential for epidemiology – Complete genome sequences (human and pathogens/vectors) – SNPs – Genotypes – Population Sets – Supplemental data for specific Journal articles

Gen. Bank is a Database • Contains all DNA and protein sequences described in the scientific literature or collected in publicly funded research • Flatfile: Composed entirely of text – you could print the whole thing out • Each submitted sequence is a record • Had fields for Organism, Date, Author, etc. • Unique identifier for each sequence – Locus and Accession #

Fields
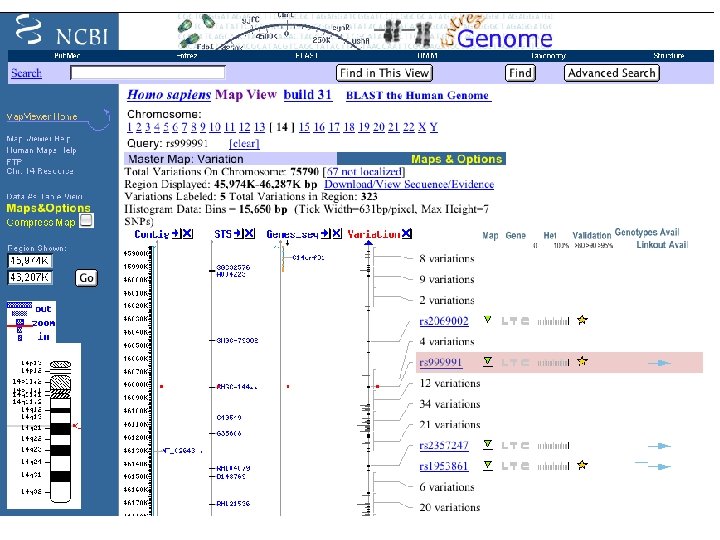
 Cluster Report: rs 1042574 Organism: human (Homo")
db. SNP record Reference SNP (ref. SNP) Cluster Report: rs 1042574 Organism: human (Homo sapiens) Molecule Type: Genomic Created/Updated in build: 86/141 Map to Genome Build: 106/Weight Validation Status: by. Cluster Variation Class: SNV: single nucleotide variation Ref. SNP Alleles: C/T (FWD) Allele Origin: Ancestral Allele: C Variation Viewer: unknown Clinical Significance: NA MAF/Minor. Allele. Count: NA MAF Source: HGVS Names: NC_000014. 9: g. 24166518 C>T, NM_006084. 4: c. *322 C>T, NT_026437. 13: g. 5942995 C>T >gnl|db. SNP|rs 1042574|allele. Pos=140|total. Len=345|taxid=9606|snpclass=1|alleles='C/T'|mol=Genomic|build=138 CCTTTTWADTTT GAGATATACG CCCTCTTTCA TCTGTAAGGG ACTAGGAAAT TCCAAATGGT GTGAACCCAG GGGGCCTTTC CCTCTTCCCT GACCTCCCAA CTCTAAAGCC AAGCACTTTA TATTTTCCT Y TTAGATATTC MCTAAGGACT TAAMATAAAA TTTTATTGAA AGAGGAATCA GTATCTGATT TTCTGGGAGA AGAAGGTAGC AGTGGTCACA GATAGAGATG TAAACTTAAG AGTGGGGCAC TGGGGTTCTC TTCCTGCTGA CATCTCCAGC CTCTTTCCTCTGCCC ACAGGTTCTG GCTAAGAKGC TGCCTGGGCC CTGTG Summary Average Individual Het. +/- std err: Count 2 Validation status Founders Count 2 Marker displays Mendelian segregation UNKNOWN Individual Overlap 0 Genotype Conflict 0 PCR results confirmed in multiple reactions UNKNOWN Homozygotes detected in individual genotype data UNKNOWN

Accession Numbers!! • Databases are designed to be searched by accession numbers (and locus IDs) • These are guaranteed to be non-redundant, accurate, and not to change. • Searching by gene names and keywords is doomed to frustration and probable failure • Neither scientists nor computers can be trusted to accurately and consistently annotate database entries • If only scientists would refer to genes by accession numbers in all published work!

http: //www. ncbi. nlm. nih. gov/Genbank • Gen. Bank is managed by the National Center for Biotechnology Information (NCBI) at the NIH (part of the U. S. National Library of Medicine) • Once upon a time, Gen. Bank mailed out sequences on CD-ROM disks a few times per year. • Now Gen. Bank is over 150 billion bases • Scientists access Gen. Bank directly over the Web at www. ncbi. nlm. nih. gov

What is Gen. Bank? Gen. Bank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences ( Nucleic Acids Research 2007 Jan ; 35(Database issue): D 21 -5). There approximately 65, 369, 091, 950 bases in 61, 132, 599 sequence records in the traditional Gen. Bank divisions and 80, 369, 977, 826 bases in 17, 960, 667 sequence records in the WGS division as of August 2006.

Relational Databases • Databases can be more complex than a single spreadsheet • Gen. Bank has proteins and SNPs as well as DNA • Some fields (i. e. phosphorylation sites) apply to protein, but not DNA • Better to create a separate spreadsheet format for Protein records • Each different spreadsheet is called a Table • Different Tables are linked by key fields – (i. e. DNA and protein for same gene)

Many Tables at NCBI • The NCBI hosts a huge interconnected database system that, in addition to DNA and protein, includes: – Journal Articles (Pub. Med) – Genetic Diseases (OMIM) – Polymorphisms (db. SNP) – Cytogenetics (CGH/SKY/FISH & CGAP) – Gene Expression (GEO) – Taxonomy – Chemistry (Pub. Chem)

Database Design A database can only be searched in ways that it was designed to be searched You can search within a specific Field in a specific Table - and sometimes can combine searches from different Fields and/or Tables (Boolean: "AND" and "OR" searches) Bad to search for "human hemoglobin" in a 'Description' field Much better to search for "homo sapiens in 'Organism' AND "HBB" in 'gene name'

Web Query • Most Scientific databases have a webbased query tool • It may be simple…

… or complex

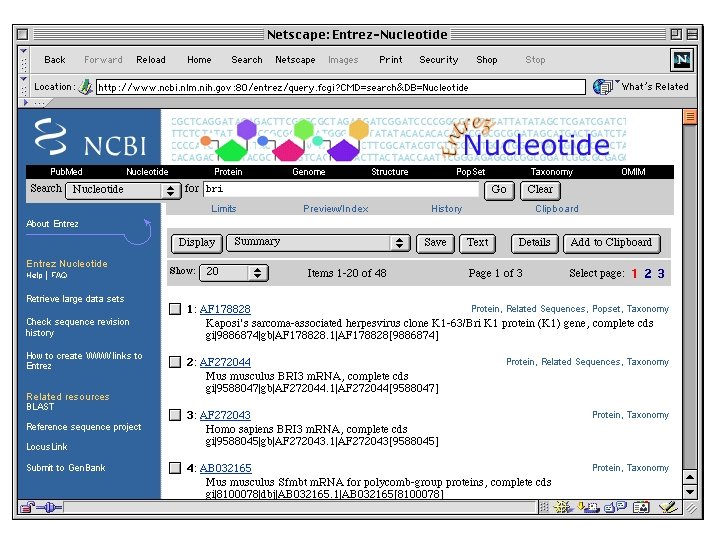
ENTREZ is the Gen. Bank web query tool

Advanced query interface:

Web API • In addition, many public databases have a specific query language that can be used by any software to create automated queries. • This is usually known as an Application Programming Interface (API). • If the interface communicates over the http protocol (used by web browsers), then it is a Web API (the simplest to work with as a novice programmer)

ENTREZ has pre-computed links between Tables • Relationships between sequences are computed with BLAST • Relationships between articles are computed with "MESH" terms (shared keywords) • Relationships between DNA and protein sequences rely on accession numbers • Relationships between sequences and Pub. Med articles rely on both shared keywords and the mention of accession numbers in the articles.

NCBI Databases contain more than just DNA & protein sequences
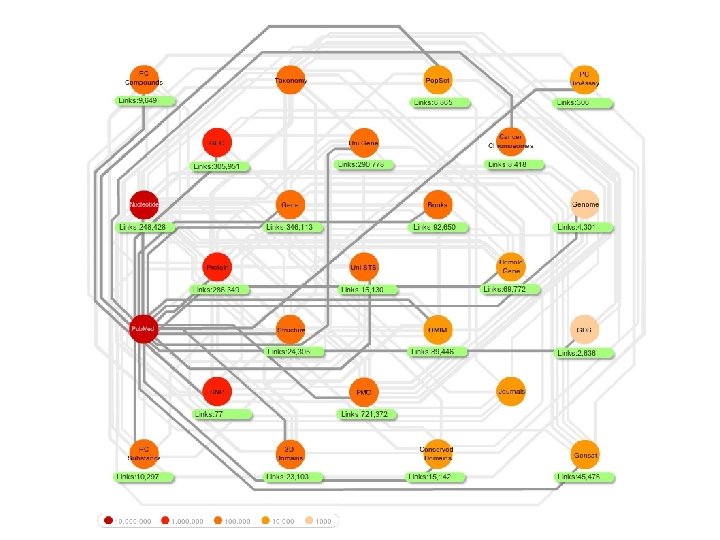

Other Important Databases • • Genomes Proteins Biochemical & Regulatory Pathways Gene Expression Genetic Variation (mutants, SNPs) Protein-Protein Interactions Gene Ontology (Biological Function)

UCSC Genome Browser Search by gene name: or by sequence:

BED format • Genome Browsers use a BED format that defines a genomic interval as positions on a reference genome. • An interval can be a anything with a location: gene, exon, binding site, region of low complexity, etc. • BED files can also specify color, width, some other formatting. chromosome start chr 1 chr 2 chr 3 213941196 213942363 213943530 158364697 158365864 127477031 127478198 127479365 end 213942363 213943530 213944697 158365864 158367031 127478198 127479365 127480532 track name="Item. RGBDemo" description="Item RGB demonstration" item. Rgb="On" chr 7 127471196 127472363 Pos 1 0 + 127471196 127472363 255, 0, 0 chr 7 127472363 127473530 Pos 2 0 + 127472363 127473530 255, 0, 0 chr 7 127473530 127474697 Pos 3 0 + 127473530 127474697 255, 0, 0 chr 7 127474697 127475864 Pos 4 0 + 127474697 127475864 255, 0, 0 chr 7 127475864 127477031 Neg 1 0 - 127475864 127477031 0, 0, 255 chr 7 127477031 127478198 Neg 2 0 - 127477031 127478198 0, 0, 255 chr 7 127478198 127479365 Neg 3 0 - 127478198 127479365 0, 0, 255 chr 7 127479365 127480532 Pos 5 0 + 127479365 127480532 255, 0, 0

http: //genome. ucsc. edu/FAQformat. html#format 1 The first three required BED fields are: 1. chrom - The name of the chromosome (e. g. chr 3, chr. Y, chr 2_random) or scaffold (e. g. scaffold 10671). 2. chrom. Start -The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0. 3. chrom. End - The ending position of the feature in the chromosome or scaffold. The chrom. End base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chrom. Start=0, chrom. End=100, and span the bases numbered 0 -99. The 9 additional optional BED fields are: 4. name - Defines the name of the BED line. This label is displayed to the left of the BED line in the Genome Browser window when the track is open to full display mode or directly to the left of the item in pack mode. 5. score - A score between 0 and 1000. If the track line use. Score attribute is set to 1 for this annotation data set, the score value will determine the level of gray in which this feature is displayed (higher numbers = darker gray). 6. strand - Defines the strand - either '+' or '-'. 7. thick. Start - The starting position at which the feature is drawn thickly (for example, the start codon in gene displays). When there is no thick part, thick. Start and thick. End are usually set to the chrom. Start position. 8. thick. End - The ending position at which the feature is drawn thickly (for example, the stop codon in gene displays). 9. item. Rgb - An RGB value of the form R, G, B (e. g. 255, 0, 0). If the track line item. Rgb attribute is set to "On", this RBG value will determine the display color of the data contained in this BED line. NOTE: It is recommended that a simple color scheme (eight colors or less) be used with this attribute to avoid overwhelming the color resources of the Genome Browser and your Internet browser. 10. block. Count - The number of blocks (exons) in the BED line. 11. block. Sizes - A comma-separated list of the block sizes. The number of items in this list should correspond to block. Count. 12. block. Starts - A comma-separated list of block starts. All of the block. Start positions should be calculated relative to chrom. Start. The number of items in this list should correspond to block. Count. In BED files with block definitions, the first block. Start value must be 0, so that the first block begins at chrom. Start. Similarly, the final block. Start position plus the final block. Size value must equal chrom. End. Blocks may not overlap. Example: Here's an example of an annotation track that uses a complete BED definition: track name=paired. Reads description="Clone Paired Reads" use. Score=1 chr 22 1000 5000 clone. A 960 + 1000 5000 0 2 567, 488, 0, 3512 chr 22 2000 6000 clone. B 900 - 2000 6000 0 2 433, 399, 0, 3601
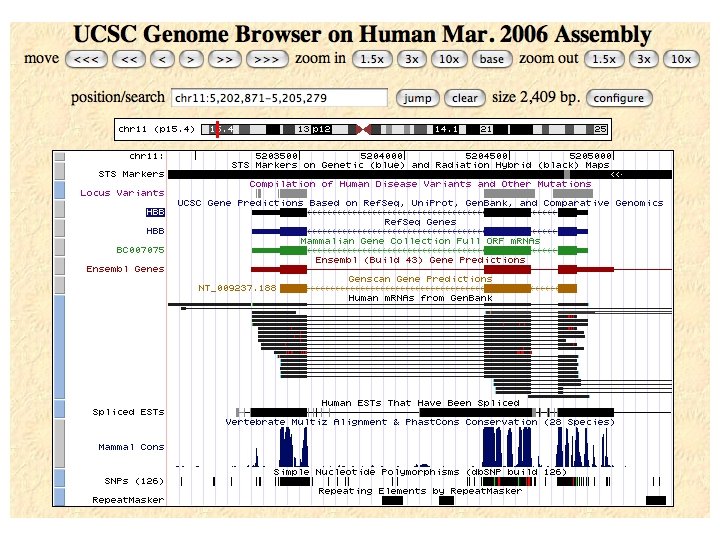

Lots of additional data can be added as optional "tracks" - anything that can be mapped to locations on the genome

Ensembl at EBI/EMBL
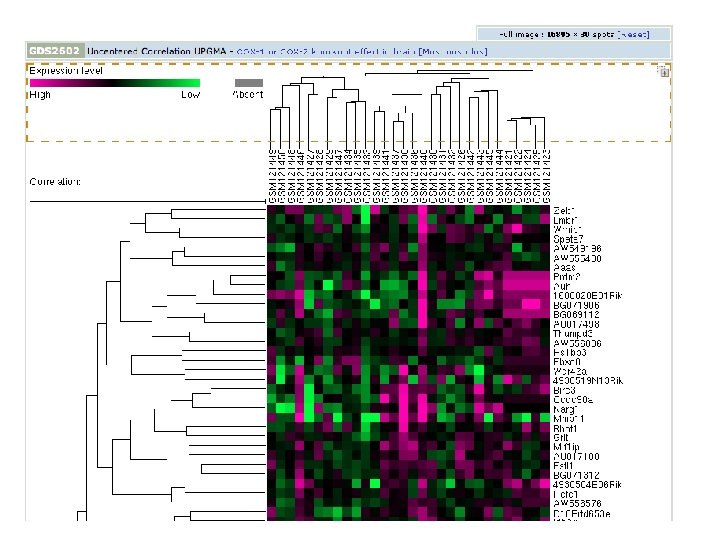
 • Genetic variation • Can be alleles of genes •")
SNPs (Single Nucleotide Polymorphisms) • Genetic variation • Can be alleles of genes • also differences in non-coding regions collected from genome sequencing of different individuals • db. SNP at the NCBI - all public SNP data • SNP Consortium at CSHL - high quality set

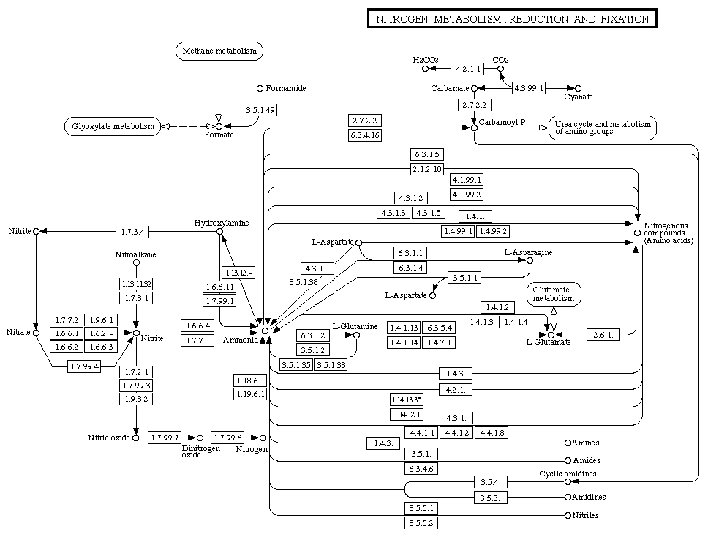
KEGG: Kyoto Encylopedia of Genes and Genomes • Enzymatic and regulatory pathways • Mapped out by EC number and crossreferenced to genes in all known organisms (wherever sequence information exits) • Parallel maps of regulatory pathways
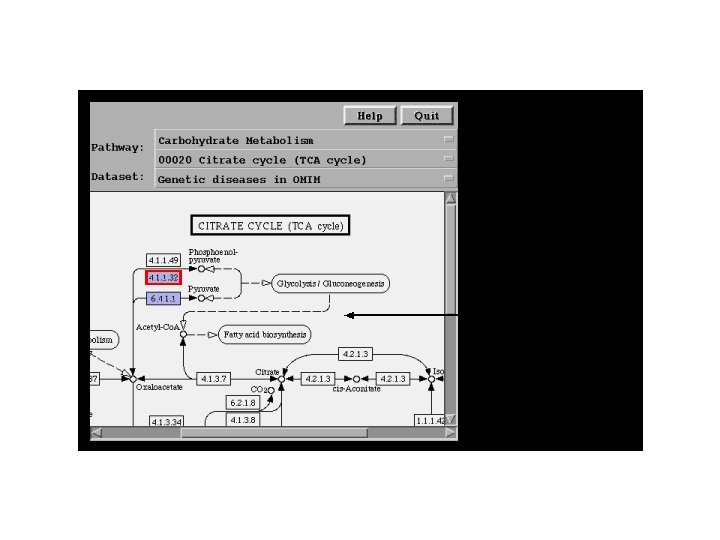
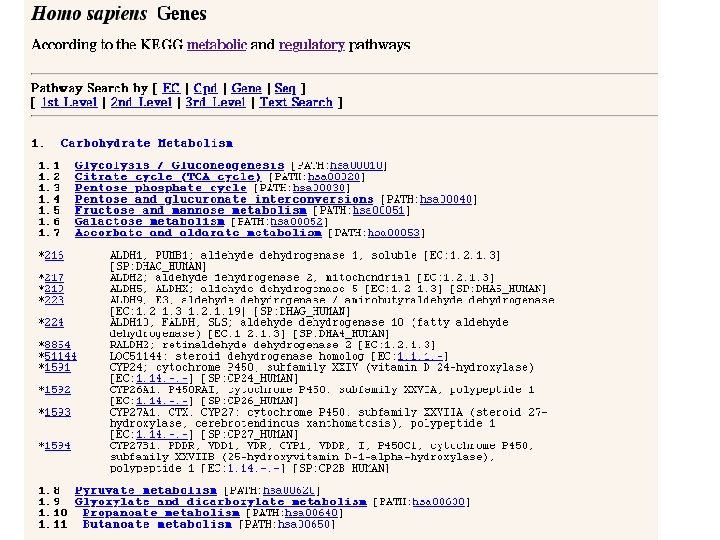

NCI Bio. Carta

Protein-Protein Interactions • • Metabolic and regulatory pathways Transcription factors Co-expression Biochemical data – crosslinking – yeast 2 -hybrid – affinity tagging • Useful feedback to genome annotation/protein function and gene expression
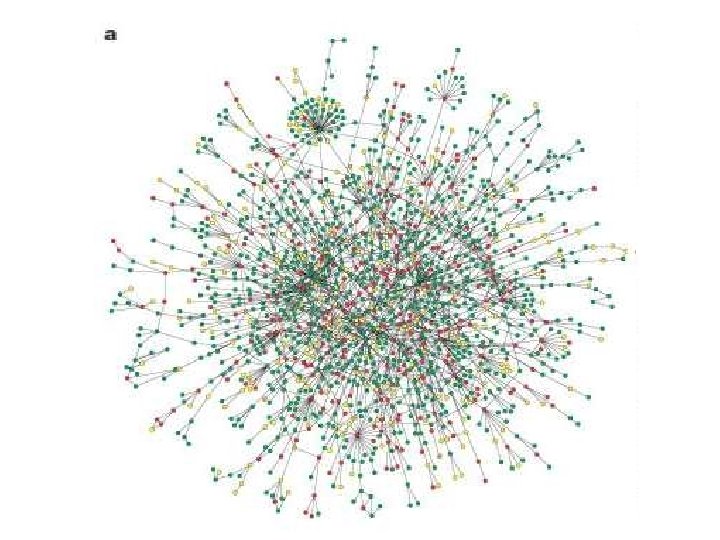

BIND - The Biomolecular Interaction Network Database

Genome Ontology • Genetics is a messy science • Scientists have been working in isolation on individual species for many years - naming genes, mutants, odd phenotypes – “sonic hedgehog” • Now that we have complete genome sequences, how to reconcile the names across all species? • Genome Ontology uses a single 3 part system – Molecular function (specific tasks) – Biological process (broad biologial goals - e. g cell division) – Cellular component (location)
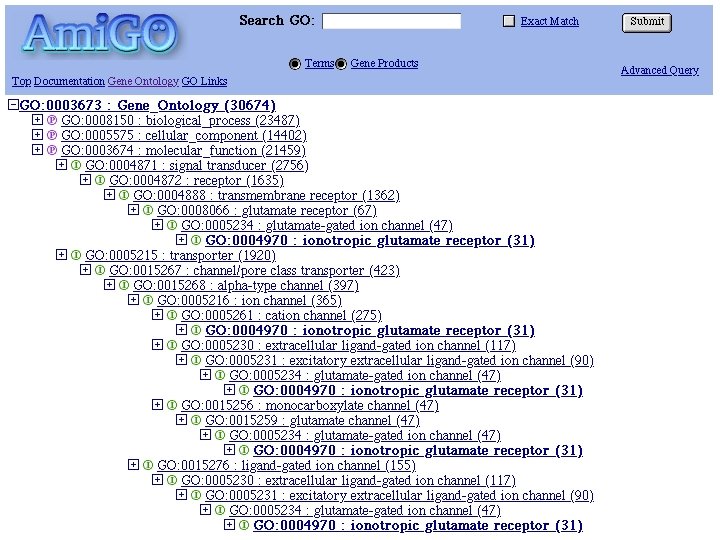

Database Search Strategies • General search principles - not limited to sequence (or to biology) • Use accession numbers whenever possible • Start with broad keywords and narrow the search using more specific terms • Try variants of spelling, numbers, etc. • Search all relevant databases • Be persistent!!

Bioinformatics Paradigm • Find the data • Download the data • Reformat the data • Collect the samples • Run molecular analysis • Filter the data • Run analysis software • Collect and sort results • Publish / Data sharing

Summary • text formats for some common genomics data types • need for universal standard data format • data types that lack a universal standard • formatting text with tag: value pairs • basic database concepts • fields, records, unique identifiers • tab and csv formats • relational databases, tables • details of the FASTA format • header + data • Data formats in public molecular biology databases • Genbank, db. SNP • Genome Browsers: BED format • Database queries: field specific queries • Genome Ontology

Next Lecture: Python I
- Slides: 66