Bioinformatics Lecture 1 DNA the basics Drew Berry

Bioinformatics Lecture 1

DNA - the basics

Drew Berry – DNA animations http: //www. youtube. com/watch? v=WFCvkk. DSf. IU&index=4&list=PL 9 CBBEA 5 A 85 DBCDEF

Organisation of DNA • DNA is packed in Chromosomes • Karyotype: chromosome set of a species • Chromosomes are dynamic structures The Human karyotype: • 23 pairs of chromosomes • 46 DNA molecules

DNA replication • The ability of DNA to replicate itself is a fundamental driver of life • DNA copy is catalysed by enzymes (DNA polymerases) • The complementary strand is synthesised from a template strand, using deoxynucleotides and a primer • Synthesis is directional (5’->3’) Deoxyribonucleotides d. NTPs A Primer Template DNA strand T DNA polymerase 5’ Template 3’ TCAG 5’ 3’ AG TC reverse complement copy C G

The polymerase chain reaction • Replication requires a DNA polymerase • Thermostable DNA polymerase (eg Taq polymerase) • Efficient DNA amplification • No error correction Melt DNA (94 -98 °) Anneal primers (50 -65 °) Kary Mullis Nobel prize in chemistry: 1993 Elongation (72 °) Exponential replication
 • PCR Reaction is terminated using randomly incorporated dideoxynucleosides (dd. NP)")
DNA Sequencing (Sanger) • PCR Reaction is terminated using randomly incorporated dideoxynucleosides (dd. NP) • Older methods use radiolabelled phosphate • Newer methods use dd. NP incorporating dyes • Truncated DNA strands are separated on a gel or by capillary electrophoresis

Next Generation Sequencing • Next generation sequencing refers to methods newer than the Sanger approach • A variety of techniques developed by different companies • DNA is generally immobilized on a solid support • Very large numbers of small reads • Multiple reads of a each section of genomic DNA (eg 30 x) • Assembling the genome becomes a significant computational problem • Some ‘single molecule’ methods do not require PCR (reduces errors) • Cost has reduced substantially the $1000 genome! • Refs: Metzker, M. L. Sequencing Technologies — the Next Generation. Nat. Rev. Genet. 2009, 11, 31– 46.

The Human Genome Project • Funded by US government • The human genome was published in February 2001 • Project completed in 2003 • Cost $US 2. 7 billion in 1991 dollars • Hierarchical shotgun sequencing (genome is broken down into many smaller fragments) • Automated Sanger type sequencing • Ref: http: //www. nature. com/scitable/topicpage/dnasequencing-technologies-key-to-the-human-828

Human genome by function • • The human genome contains about 21 K genes (about 100, 000 were expected!) 98% of the human genome is noncoding DNA Noncoding DNA can code for regulatory RNAs or otherwise regulate transcription Ref: Häggström, Wikiversity Journal of Medicine 1 (2). DOI: 10. 15347/wjm/2014. 008. ISSN 20018762

The druggable genome – Current drug targets Ref: Hopkins, A. L. ; Groom, C. R. The Druggable Genome. Nat Rev Drug Discov 2002, 1, 727– 730.

The druggable genome – Human genes Ref: Hopkins, A. L. ; Groom, C. R. The Druggable Genome. Nat Rev Drug Discov 2002, 1, 727– 730.

Human genome resources • Three useful sites providing a huge number of resources such as genome browsers • NCBI: National center of biological information – http: //www. ncbi. nlm. nih. gov/genome/guide/human/ • UCSC genome browser – http: //genome. ucsc. edu/ • Ensembl: European site at the Sanger centre – http: //www. ensembl. org

Next-gen Sequencing Overview • Ref: http: //res. illumina. com/documents/products/illumina_sequencing_introduction. pdf

Multiple Genomes • Ref: Mc. Vean et al. An Integrated Map of Genetic Variation From 1, 092 Human Genomes. Nature 2012, 491, 56 -65.

Bioinformatics • Sequencing technologies produce enormous amounts of sequence data. What do we want to do with this? – Identify genes – Identify functions of gene products (proteins) – Compare genes between species – Identify relationships (similarities) between species

The Genetic Code In general: • Amino acids that share the same biosynthetic pathway tend to have the same first base in their codons • Amino acids with similar physical properties have similar codons causing conservative substitutions in the case of mutations or mistranslation

Genetic mutation The genetic code can be changed by a variety of processes Small scale: • Damage to DNA (radiation or chemical damage) • Translation errors Large scale: • Duplication of sections of DNA • Deletion of sections of DNA • Transposition of sections of DNA
")
The rate of genetic mutation • The mutation rate (per year or per generation) differs between species and even between different sections of the genome • Different types of mutations occur with different frequencies • The average mutation rate is estimated to be ~2. 5 × 10− 8 mutations per nucleotide site or 175 mutations per diploid genome per generation • Ref: Nachman, M. W. ; Crowell, S. L. Estimate of the Mutation Rate Per Nucleotide in Humans. Genetics, 156, 297 (2000).

Amino acid substitution matrices • Substitution matrices describe the probability that one AA is converted to another and ‘accepted’ • Matrix is a ‘log odds’ matrix – i. e. here the probability of conversion from Ala to Arg is 1/log(30)

PAM and BLOSUM matrices • Scoring matrices are used to: – produce sequence alignments and score similarity between two or more protein – to search a database to find sequences similar to a test sequence • Commonly used families of matrices: – PAM (Accepted Point Mutation) matrices (Dayhof) • Derived from global alignments of entire proteins • Better for closely related protens – BLOSUM (BLocks SUbstitution Matrices) matrices (Steven and Henikof) • Derived from local alignments of blocks of sequences • Better for evolutionally divergent sequences

BLAST - Searching genomes • BLAST is a rapid method for searching protein or DNA sequences in large databases • Sequences are divided into groups k AAs or Bases PGFHJIQMQVVS PGF, GFH, FHJ, HJI, etc (k=3) • Common or repeated sequences are discarded • Sections of exact sequence match are searched for • The sequence alignment is expanded from sections that are exact matches • Blast can miss difficult matches

http: //blast. ncbi. nlm. nih. gov/
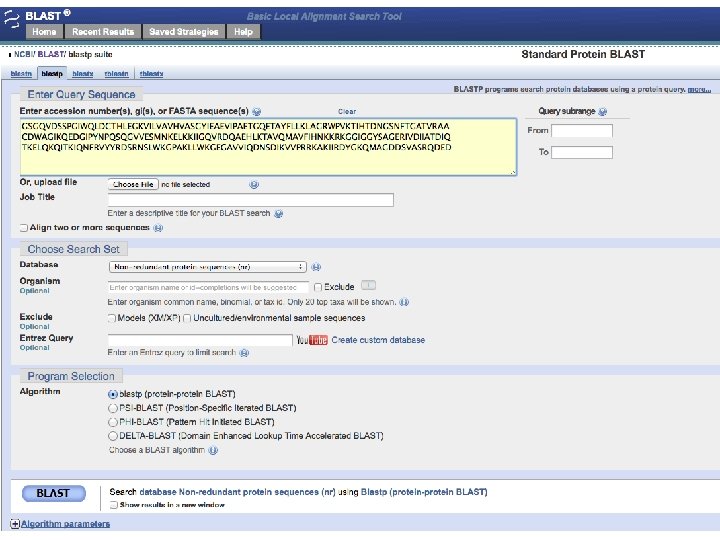
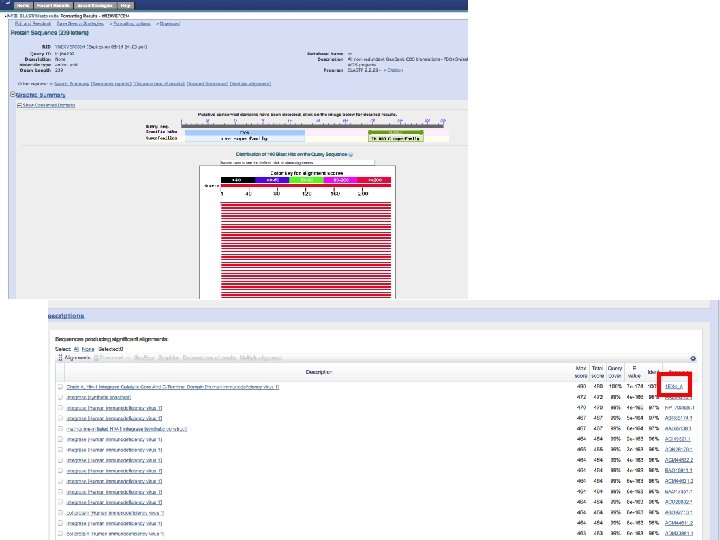

Sequence alignment • Protein or DNA sequences can be aligned • Differences between sequences are interpreted as mutations, insertions or deletions • Substitution matrices are used to score the likelihood of a match • Alignment scores are calculated between pairs of sequences • Multiple alignments can be performed • Many alignment programs: Clustal, T-coffee,

Clustal

Sequence alignments and protein structural similarity • Sequence alignments are based on protein/DNA sequence similarity and not on structural similarity • High sequence similarity implies (but does not guarantee) structural similarity • High sequence similarity implies (but does not garuantee) similar protein function Comparison of RMSD when pairs of similar proteins are superimposed using the sequence alignment (X axis) and the protein 3 D structures (Y axis) Ref: Kosloff, M. ; Kolodny, R. Sequence-Similar, Structure-Dissimilar Protein Pairs in the PDB. Proteins 2008, 71, 891

Differences between sequence and structural alignment Chain A versus chain D from PDB ID 1 vr 4. The two chains are 100% identical in sequence A: Alignment by sequence B: Alignment by structure C: Overlaid structures Ref: Kosloff, M. ; Kolodny, R. Sequence-Similar, Structure-Dissimilar Protein Pairs in the PDB. Proteins 2008, 71, 891

Improving sequence alignments • Adding structural information to sequence alignments can improve their quality

Summary • This lecture should provide an overview of: • • DNA sequencing and the Polymerase Chain Reaction Genome sequencing BLAST searching Sequence alignments and their limitations
- Slides: 31