Interstitial Lung Diseases Diffuse Parenchymal Lung Diseases DPLD
")


 and/or")



 2.")





. 3. Stop smoking. 4. Annual pneumococcal vaccines.")


 or abnormal liver function tests")




- Slides: 23
Interstitial Lung Diseases Diffuse Parenchymal Lung Diseases (DPLD)
Case-studies A 65 -year-old man presents with progressive shortness-of-breath. On examination he is found to have fine crackles in both lung bases and oxygen saturations of 93% on room air. A diagnosis of idiopathic pulmonary fibrosis is suspected. Which one of the following chest x-ray findings develops first in patients with idiopathic pulmonary fibrosis? A/ Asymmetrical upper zone 'ground-glass' changes B/ Small, peripheral opacities in the lower zones C/ Perihilar horizontal septal lines D/ Honeycombing E/ Loss of left heart border
A 68 -year-old male patient presents with a 6 -month history of shortness of breath that is worse on exertion with reduced exercise tolerance & no associated wheeze, or haemoptysis but does have a dry cough. He has hypertension and takes amlodipine 5 mg once a day. He has never smoked or worked with asbestos On examination, he is comfortable at rest with oxygen saturations 95% on air. There is no evidence of lymphadenopathy, clubbing or cyanosis. He has fine crackles at both lung bases that do not alter on coughing. Which of the following investigation findings would support a diagnosis of idiopathic pulmonary fibrosis? A/ Reticular changes on CT imaging that is worse at the bases B/ Obstructive picture on spirometry C/ Extensive ground glass opacities on CT imaging D/ Increased transfer factor on spirometry E/ A lymphocytosis on bronchoalveolar lavage
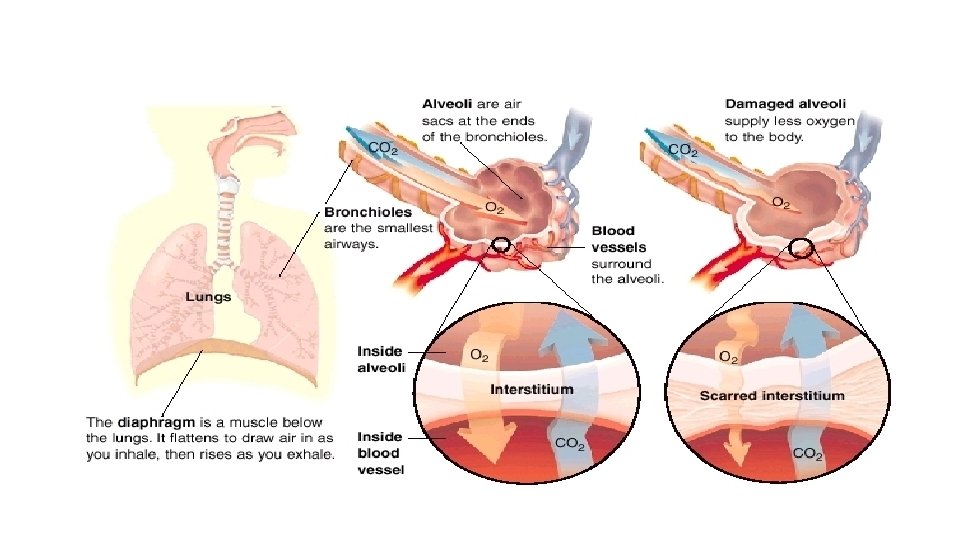
DPLD - indicates a group of diseases affecting the lung interstitium (parenchymal tissue) and/or alveolar lumen and share common clinical & radiological features.
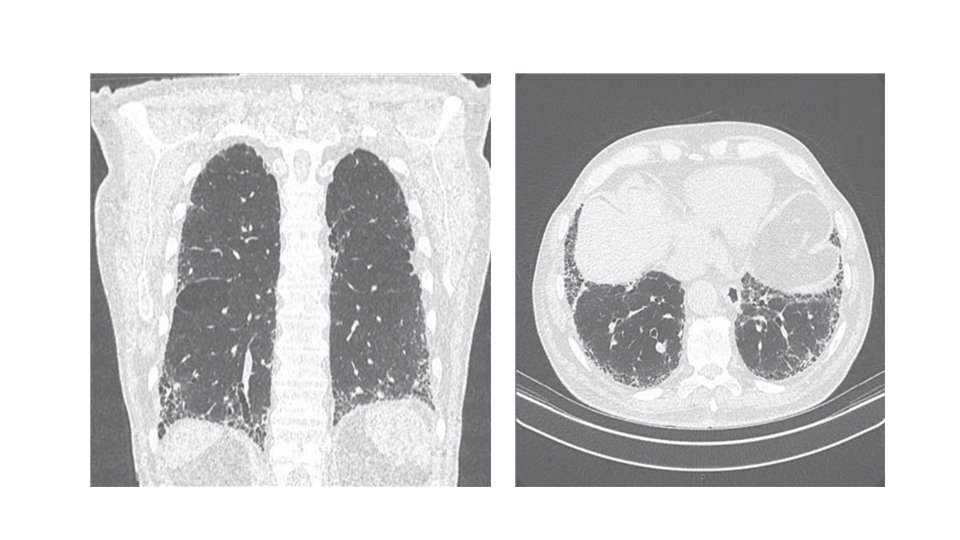
Symptoms & signs: persistent dry cough, insidious and progressive shortness of breath, inspiratory crackles & digital clubbing. Radiology: CXR- small lung volume, reticulonodular shadowing. High-resolution CT- (the diagnostic test of choice) showing ground-glass appearances, reticulonodular shadowing, honeycomb cyst & traction bronchiectasis. Pulmonary function test: restrictive ventilatory defect, reduced lung volume & decrease gas exchange.
Classification 1. Idiopathic interstitial pneumonia. 2. DPLD of known cause, (e. g. drugs or association with connective tissue disease). 3. Granulomatous DPLD, (e. g. sarcoidosis). 4. Other forms of DPLD, (e. g. lymphangioleiomyomatosis, histiocytosis X etc).
Idiopathic interstitial pneumonias are of unknown cause & usually classified according to the histological features on bronchial biopsy into: A/Idiopathic pulmonary fibrosis. B/ Idiopathic interstitial pneumonia other than idiopathic pulmonary fibrosis; including: 1. Desquamative interstitial pneumonia. 2. Respiratory bronchiolitis interstitial lung disease. 3. Acute interstitial pneumonia. 4. Cryptogenic organising pneumonia (‘bronchiolitis obliterans organising pneumonia’ – BOOP). 5. Non-specific interstitial pneumonia. 6. Lymphocytic interstitial pneumonia.
Differential diagnosis of DPLD 1. Infection (viral pneumonia, pneumocystis jirovecii, mycoplasma pneumoniae, tuberculosis) 2. Malignancy (Leukaemia and lymphoma, lymphangitic carcinomatosis, multiple metastases, bronchoalveolar carcinoma). 3. Pulmonary oedema. 4. Aspiration pneumonitis.
Idiopathic pulmonary fibrosis Progressive fibrosing interstitial pneumonia in the adults of unknown aetiology with histological (alveolar epithelium focal damage) & radiological pattern of usual interstitial pneumonia. • IPF has strong association with smoking. • Familial disease is rare but genetic role can be significant. DDx: lung fibrosis secondary to drugs, occupational exposure or connective tissue diseases.
Clinical presentation: • Uncommon before the age of 50 years. • Progressive insidious breathlessness with dry cough. • Bilateral fine basal end-inspiratory crackles & digital clubbing. • Patients can develop exacerbations during the chronic course of the disease with severe SOB, reduced gas transfer & new ground-glass changes or consolidations on chest CT.
Investigations: A/ CXR: bilateral lower lobe and subpleural reticular shadowing but can be normal in early or limited disease. B/ High-Resolution CT of the chest (HRCT): patchy, predominantly peripheral, subpleural and basal reticular shadows. Honeycombing cysts and traction bronchiectasis in advanced disease. • It can be useful to exclude other differential diagnoses such as hypersensitivity pneumonitis, sarcoidosis and asbestosis
C/ Pulmonary function test: restrictive defect with reduced lung volumes and gas transfer. • Hypoxemia & hypocapnia initially presents with exercise but later on at rest as the disease progressed. D/ Bronchoscopy & lung biopsy: only indicated in uncertain cases that cannot be diagnosed based on other tests. E/ Immunology: antinuclear antibody (ANA) or anti-cyclic citrullinated peptide 2 (anti-CCP 2) may be mildly positive.
Management: Goal of management is to maintain vital capacity between 50% to 80% of predicted. However, there 1. Disease-modifying therapy: used to maintain lung and prevent further deterioration with either the antifibrotic agent (pirfenidone) or the tyrosine-kinase inhibitor (nintedanib). However, no symptomatic benefit (cough & dyspnoea do not change) is obtained with this treatment. • If patient shows 10% deterioration in lung function during the first year of treatment, disease-modifying therapy should be discontinued. • Side effects include photosensitivity with pirfenidone and diarrhoea with nintedanib. • Oral N‑acetylcysteine can be used in the management of IPF but its benefits are uncertain.
2. Treatment of gastro-oesophageal reflex (relieving cough). 3. Stop smoking. 4. Annual pneumococcal vaccines. 5. Pulmonary rehabilitation program & domiciliary O 2 therapy in severe cases. 6. Lung transplantation (in severe cases). • In acute exacerbations, the treatment mainly supportive (antibiotics, glucocorticoids, immunosuppression & respiratory support).
Prognosis • Prognosis is poor with 3 -years survival is the expected outcome. However, cases with minimal disease can live longer. • Progression of the disease results in central cyanosis with pulmonary hypertension & right side heart failure. • Predictors of poor prognosis are serial pulmonary function tests showing reduced gas transfer & O 2 desaturation during exercise & lung biopsy indicating presence of fibroblastic foci.
Sarcoidosis is a multisystem granulomatous disorder of unknown aetiology that is characterised by the presence of non-caseating granulomas. • More common in northern Europe.
Clinical features: • Asymptomatic: abnormal routine chest X-ray (~30%) or abnormal liver function tests • Respiratory and constitutional symptoms (20– 30%) • Erythema nodosum and arthralgia (20– 30%) • Ocular symptoms (5– 10%) • Skin sarcoid (including lupus pernio) (5%) • Superficial lymphadenopathy (5%) • Other (1%), e. g. hypercalcaemia, diabetes insipidus, cranial nerve palsies, cardiac arrhythmias, nephrocalcinosis
Löfgren’s syndrome – an acute illness characterised by erythema nodosum, peripheral arthropathy, uveitis, bilateral hilar lymphadenopathy (BHL), lethargy and occasionally fever – is often seen in young women.
Investigations: 1. Blood tests: lymphopenia, mild impairment of LFT, hypercalcaemia, Hypercalciuria. 2. CXR: bilateral hilar lymphadenopathy, pulmonary infiltrates & fibrosis. 3. Bronchoscopy: biopsy showing cobblestone mucosal appearance & noncaseating granulomas.
Management: • Most patients recover spontaneously. • Treatment includes NSAIDs, steroids and immunosuppressive therapy in advanced cases.
Thank you