Interstitial Lung Disease Huda Etoom Restrictive lung disease


 chest")




 pattern: 1)Bilateral subpleural reticular changes most prominent in lower, posterior lung")
 UIP: subpleural reticulation associated with honeycomb changes and fibroblast")
 antifibrotic therapy (pirfenidone and nintedanib) can slow decline of lung function in")


 Diffuse peripheral subpleural, symmetric, ground glass, and reticular opacities are")


respiratory bronchiolitis")










Wegener’s disease, • Rare with unkown cause • Charecterized by necrotizing granulomatous vasculitis that")
SARCOIDOSIS • Most common granulomatous disease • Immune mediated widespread noncaseating granuloma • Elevated")

HISTOCYTOSIS X pulmonary Langerhans cell histiocytosis X (PLCH) • Chronic interstitial pneumonia caused")
 Hypersenitivity pneumonitis: Inhalation of")





 Clinical Manifestations • AIP is a")


")

- Slides: 43
Interstitial Lung Disease Huda Etoom
Restrictive lung disease: Lung is restricted from air filling, air cant get in restricted lung expantion cause decrease lung volume and capacity (fev 1 and tlc) pfts: increase or normal fev 1fvc ratio(fev 1 decreased proportionally to fvc) Loop is shifted to the right Severity? The lower the tlc the more sever the disease
Types : Intrinic: : Parynchemal, Ild Extrinsic: : Poor breathing mechanism (extrapulmonary causes) chest wall: poor muscular effort , poor structural apparatus, pleura; effusion From the abdomen: ascites Diaphragm, paralysis
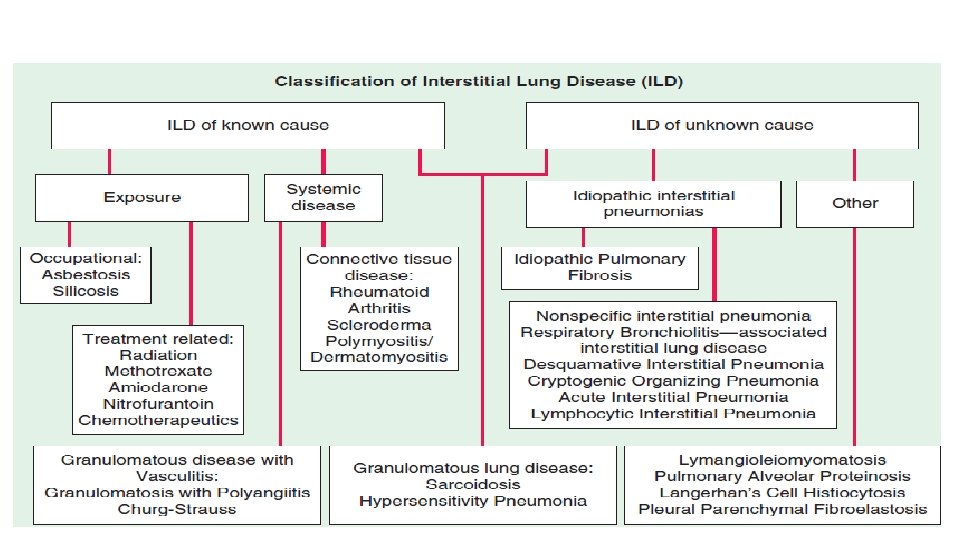
• Ild: is defined as an inflammatory process involving the alveolar wall resulting in widespread fibroelastic proliferation and collagen deposition that can lead to irreversible fibrosis, distortion of lung architecture and impaire gas exchange. • The term interstitial is misleading since most also associated with extensive alteration of alveolar and airway architecture. • include a large number (>200) of heterogeneous conditions that affect the lung parenchyma with varying degrees of inflammation and fibrosis. • The prognosis is variable and depend on the diagnosis. • Ild can be associated with high morbidity and mortality. • IPF, sarcoidosis, and ILDs related to CTDs most common forms of ILD.
complaint: • most patients ultimately diagnosed with an ILD will come to medical attention with reports of progressive exertional dyspnea(short shallow brathing) or a persistent dry cough. • because some ILDs are part of multisystem disorders, some patients will be identified based on non-respiratory symptomatology (e. g. , skin thickening in the setting of systemic sclerosis) • ILDs can also be identified incidentally based on investigation
IDIOPATHIC INTERSTITIAL PNEUMONIAS 1**IDIOPATHIC PULMONARY FIBROSIS • IPF is the most common ILD of unknown cause. • Prevalence increases with age • IPF is commonly diagnosed in the fifth or sixth decade in life, unusual in young adult. • affects men more than women • Exposure history: idiopathic but frequently associated with a history of smoking or other environmental exposures. • Gradual in onset • Genetic finding may explain more than third of the risk of the disease
• Clinical symptoms: Gradual onset of sob, dry cough • Examination: Frequent rales at lung base Digital clubbing is common • Prognosis: IPF is a variably progressive disease that carries a poor prognosis with an estimated 50% 3– 5 -year survival.
UIP(unusual interstitial pattern) pattern: 1)Bilateral subpleural reticular changes most prominent in lower, posterior lung zones. 2)Traction bronchiectasis 3)honeycombing common. Classic UIP pattern is considered diagnostic.
• Histopathology (definitive diagnosis) UIP: subpleural reticulation associated with honeycomb changes and fibroblast foci **These fibrotic changes alternate with areas of preserved normal alveolar architecture consistent with temporal and spatial heterogeneity(temporal and spatial heterogenicity)
Treatment 1) antifibrotic therapy (pirfenidone and nintedanib) can slow decline of lung function in IPF patient may also improve survival. 2)Physical therapy and supplemental oxygen, when indicated, can improve exercise tolerance and reduce likelihood of developing pulmonary hypertension. 3)Lung transplantation can extend survival and improve the quality of life in a subset of IPF patients who meet criteria to undergo transplant **immunosuppression, which had been commonly prescribed to many IPF patients, has now been demonstrated to be associated with increased morbidity and mortality.
2**NON-SPECIFIC INTERSTITIAL PNEUMONIA *can be idiopathic NISP *NSIP is also commonly observed in patients with connective tissue disease(Positive serologic tests for connective tissue disease are frequently observed) and less frequently with familial interstitial pneumonia, drug toxicity, and infectin *commonly diagnosed in non-smoking females in their fifth decade of life. . Sympotms: subacute onset of sob and dry cough frequently assosciated with other condition. Exam: frequent rales, clubbing less common Prognosis: Idiopathic NSIP has a relatively good prognosis, with a 5 -year survival >80%; patients with a predominant cellular NSIP pattern have a more favorable prognosis than those with a fibrosing NSIP pattern.
HRCT Image Findings(not diagnostic) Diffuse peripheral subpleural, symmetric, ground glass, and reticular opacities are common. Volume loss and traction bronchiectasis involving the lower lung zones can also be found. Occasionally subpleural sparing is noted, while peribronchiolar thickening and honeycombing are uncommon.
Histopathology Diagnostic lung biopsy amounts of interstitial inflammation and fibrosis with a uniform appearance. **NSIP is often referred to histopathologically as being either predominantly cellular (and potentially more responsive to medical therapy) or fibrotic (and potentially less likely to resolve with medical therapy).
Treatment *Pulmonary fibrosis associated with connective tissue disease is commonly treated with immunosuppression *Idiopathic NSIP is often treated with oral steroids (prednisone), cytotoxic agents (mycophenolate, azathioprine, and cyclophosphamide), or biologics (rituximab). *Oxygen therapy, pulmonary rehabilitation, and lung transplantation may be required in patients with progressive disease.
3**SMOKING RELATED ILD Smoking can lead to obstrective or restrictive lung disease 1)respiratory bronchiolitis with interstitial lung disease (RB-ILD) 2) Desquamative interstitial pneumonia (DIP) Commonly felt to be the result of active or prior tobacco smoke exposure. Clinical Manifestations: *Can be asymptomatic and incedintly find *These disorders predominantly occur in active or prior tobacco smoke exposure, typically between 40 and 50 years of age. *dyspnea and cough are relatively common and symptomatic *wheezing is not rare rales is common clubbing is rare *The prevalence account for <10% *25% 7 year mortality.
• Hrct finding: Diffuse patchy centrolobular ground glass nodule Histopathology: Respiratory bronchiolitis with adjacent inflammatory and fibrosing changes. pigment laden macrophags.
4**CRYPTOGENIC ORGANIZING PNEUMONIA Clinical Manifestations: • COP typically involves patients in their 50– 60 s • often presents as a subacute flu-like illness, with cough, dyspnea, fever, and fatigue. Inspiratory rales are often present • It is commonly mistaken for pneumonia • on examination and most patients are noted to have restrictive lung deficits on pulmonary function testing with hypoxemia. • this syndrome can occur in isolation or can be secondary to an underlying connective tissue disease (e. g. , polymyositis), medications, or can result from an underlying malignancy.
HRCT Image Findings Similar to infectious pneumonia The most common findings include bilateral patchy infiltrate, sometimes migratory, subpleural consolidative opacities often with associated ground-glass opacities. Histopathology patchy regions of organizing pneumonia with granulation tissue that commonly involves the small airways, alveolar ducts, and alveoli with surrounding inflammation that can involve the alveolar walls
Treatment: NO RESPONSIVENESS TO ANTIBIOTIC Spontaneous recovery may occur *Corticosteroids usually need to be continued for at least 6 months as relapse rates are high. *Alternate cytotoxic (e. g. , mycophenolate, cyclophosphamide) or biologic (e. g. , rituximab) therapies can be helpful in both treating the disease and reducing the need for steroids. *In some patients with secondary forms of the disease, long-term therapy may be needed.
ILD ASSOCIATED WITH CONNECTIVE TISSUE DISEASE 1**SYSTEMIC SCLEROSIS • ILD is the most common pulmonary manifestation of SSc. 50% • Pulmonary hypertension can occur separately or concomitantly with ILD and is more frequent in patients with limited SSc. HRCT Image Findings Similar imaging findings noted in both patients with NSIP and IPF can be present, although findings consistent with COP and DAD may also be present. Chest –x-ray: reticular pattern of linear, nodular and line nodular densities "lower two –thirds
Histopathology changes commonly noted in patients with NSIP and IPF are frequently identified. Additionally, aspiration related to esophageal dysmotility is common in SSc, Treatment Cyclophosphamide : preservation of lung function and is associated with significant toxicity. Mycophenolate has recently been shown to have similar efficacy and improved tolerability. antifibrotic therapies (pirfenidone and nintedanib) are presently being conducted. Minimizing the risk of reflux by using high-dose proton pump inhibitors or antireflux surgery should be considered in SSc with progressive ILD. Lung transplantation can potentially be offered to select patients without significant aspiration or chest wall restriction.
■■RHEUMATOID ARTHRITIS • Clinical Manifestations: • A common extraarticular complication of RA is ILD. • Although RA is more common in females, RA-ILD is more frequent in males and in patients with a history of tobacco exposure. In a small subset of patients, ILD is the first disease manifestation of RA. • nearly 10% of the RA population; • HRCT Image Findings The most common imaging pattern of ILD in patients with RA is a UIP pattern • survival in patients with RA is decreased in those with a UIP pattern and among those with more extensive fibrosis in general.
• Histopathology • Histopathologic findings of UIP and NSIP are common. • ITreatmentn • immune suppression in RA-ILD. • immunosuppresive (e. g. , prednisone) • cytotoxic (e. g. , mycophenolate, azathioprine, cyclophosphamide, • and calcineurin inhibitors) agents have been used with variable • success. • antifibrotic therapies (pirfenidone and nintedanib) are presently being conducted. • Lung transplantation if not responsive to medical therapy.
■■DERMATOMYOSITIS/POLYMYOSITIS Clinical Manifestations ILD is present in up to 45% of patients with positive antisynthetase antibodies. • HRCT Image Findings include those consistent with NSIP with or without evidence for COP. A UIP pattern can also occur. • Histopathology The antisynthetase syndrome is associated with multiple histopathologic subtypes including NSIP, COP, and UIP. DAD, • Treatment Immunosuppresive (e. g. , prednisone) and cytotoxic (e. g. , mycophenolate, azathioprine, cyclophosphamide, and calcineurin inhibitors) agents are often used in patients with progressive ILD. In small studies relapses have been more common in patients treated with prednisone alone. • Patients who fail immune suppressive therapy can benefit from lung transplantation.
GRANULOMATOUS • Granulomatous Vasculitides • characterized by blood vessels with inflammatory infiltrates associated granulomatous lesions with or without the presence of tissue necrosis. • The lungs are commonly involved and a unique feature of these disorders is that hemoptysis can be a presenting symptom. • Although laboratory testing is often helpful and can provide specific information, biopsies of involved tissue can be essential for making the diagnosis. Many of these disorders include additional systemic manifestations. •
1)Wegener’s disease, • Rare with unkown cause • Charecterized by necrotizing granulomatous vasculitis that commonly affects the lung (including inflammatory infiltrates in small to medium sized vessels), the ears, nose, throat, and kidney (resulting in glomerulonephritis). • Common imaging abnormalities of GPA include nodules, patchy ground glass, and consolidative opacities that can be migratory, and hilar lymphadenopathy. • Positive for canca • Treatment : immunosuppressive agent and glucocoryicoid 2)Eosinophilic GPA (Churg-Strauss syndrome) • granulomatous vasculitis that affects the lung (including eosinophilic infiltrates in small to medium sized vessels) • result in numerous clinical manifestations but frequently includes chronic sinusitis, asthma, and peripheral blood eosinophilia, rash. • Systemic vasculitis result in skin, muscle, and nerve lesion • Positive for p-anca • Common imaging abnormalities of EG include peripheral consolidative opacities that can be migratory and small pleural effusions. • Treat with systemic glucocorticoid
3)SARCOIDOSIS • Most common granulomatous disease • Immune mediated widespread noncaseating granuloma • Elevated ACE and cd 4cd 8 ratio in bronchoalveolar lavage fluid • More common on African American females less than 40 • Often asymptomatic except for enlarged lymph node • Cxr show bilateral adenopathy and course reticular opacity • Ct of the chest better demonstrate the hilar and mediastinal adenopathy
• Associated with : • Bell palsy • Uveitis • Granulomas NON caseating • Lupus pernio • Erythema nodosum • Rheumatoid arithritis like arthropathy • Hypercalcemia • Treatment: steroid if symptomatic
4)HISTOCYTOSIS X pulmonary Langerhans cell histiocytosis X (PLCH) • Chronic interstitial pneumonia caused by abnormal proliferation of histiocytes (related to Langerhans cells of the skin). • Most patients (90%) are cigarette smokers. • Variants of disease include eosinophilic granuloma (localized to bone or lung), and two systemic forms—Letterer–Siwe disease and Hand–Schüller–Christian syndrome. • Common findings include dyspnea and nonproductive cough. • Other possible manifestations are spontaneous pneumothorax, lytic bone lesions, and diabetes insipidus. • CXR has a honeycomb appearance, and CT scan shows cystic lesions. • The prognosis and course are highly variable. • Traetment: Corticosteroids are sometimes effective. Lung transplantation may be necessary.
OCUPATIONAL AND ENVIRONMENTAL LUNG DISEASE: Ild ass with hyoersensitivity: 1) Hypersenitivity pneumonitis: Inhalation of an antigenic agent to the alveolar level induce immune mediated pneumonitis and chronic exposure to birds and often farmer, Acute form has flu like symptoms Reversible in early stages Cause dyspnea, cough, chest tightness, headache 2)Eosinophilic pneumonia
PNEUMOCONIOSIS • COAL WORKER PNEUMOCONIOSISBLACK LUNG DISEASE: **inhalation of coal dust that contain carbon and silica. Most have simple coal workers pneumoconiosis, wich usually cause no significant rs disability Some will develop complication which is charectarized by fibrosis prolonged exposure macrophages laden with carbon inflammation and fibrosis Its affect the upper lobes Increase risk for caplan syndrome(rheumatoid arithritis with pnemoconiois with intrapulmonary nodules) Small nodular opacities seen on imaging
• Asbestosis: Inhalation of asbestosis fibers lead to diffuse ild, predilection for lower lobes. Develop insidently many years after exposure(more than 15 to 20 yr) Increase the risk for bronchogenic carcinoma and malignant mesothelioma Increase risk for caplan syndrome Associated with shipbuilding, roofing, plumbing. Ivory white calcified supradiaphragmatic and pleural plaques are pathognomic for asbestosis Asbestosis bodies(ferrugin ou )found in alveolar sputum sample
• Silicosis: Localized and nodular parabronchial fibrosis, upper lobe Can be acute due to massive exposure leading to rapid onset and death or chronic due to prolonged exposure Increase risk for tb(its may distrupt phagolysosomes and impare macrophages) , ca , cor pulmonaleand caplan syndrome Associated with sandblasting, foundries, mines. Egg shell calcification of hilar lymph nodes on cxr
• Berylliosis: • Affects upper lobes Has acute and chronic forms Expsure to beryllium in aerospace and manufacturing idustiries Acute caused by massive exposure Chronic is very similar to sarcoidosis granuloma skin lesion and hyoercalcemia Non caseating granuloma on hitology and therefor its responsive for steroid Increase risk for cancer and cor pulmonale
DRUD RALATED LUNG FIBROSIS • Methotrexate • Amiodarone • Nitrofurantoin • Bleomycin • Busulfan • Chemotheraoy • Illicit drugs • phenytoin
ACUTE OR SUBACUTE IIPS ■■ACUTE INTERSTITIAL PNEUMONIA (HAMMAN-RICH) Clinical Manifestations • AIP is a rare and often fatal lung disorder that is characterized by an acute onset of respiratory distress and hypoxemia. • A prodromal period of symptoms : acute upper respiratory infection is common. • The mortality rate within 6 months of presentation can be quite high (>50%) and recurrences are common. • In those that recover, lung function improvement can be substantial. • AIP can be difficult to distinguish from acute respiratory distress syndrome (ARDS) and an acute exacerbation of an unsuspected underlying pulmonary fibrotic process. • HRCT Image Findings The most common imaging findings are patchy bilateral ground-glass opacities. Dependent regions of air-space consolidation are also common.
Histopathology Similar to ARDS and acute exacerbations of underlying pulmonary fibrosis, AIP presents histopathologically as diffuse alveolar damage (DAD) demonstrated Treatment is mostly supportive and often includes mechanical ventilation. There is no proven drug therapy for AIP. • Glucocorticoids are often given but they are not clearly effective and • have been demonstrated not to be beneficial in other forms of DAD
ACUTE EXACERBATIONS OF IIPS Clinical Manifestations an accelerated phase of lung injury that can occur in any ILD resulting in pulmonary fibrosis. most severe in, patients with known IPF. • Acute exacerbations are characterized by an acute onset (<30 days) of respiratory distress and hypoxemia occurring in a patient with underlying pulmonary fibrosis not explained by an alternate cause • mortality rates are very high (>85%) and mean survival periods range from as little as days to months. HRCT Image Findings The most common imaging findings include patchy bilateral groundglass opacities and dependent regions of air-space consolidation.
Histopathology DAD Treatment is mostly supportive evidence that drug therapy (e. g. , Nintedanib) may reduce the rate of acute exacerbations in patients with IPF. Immunosuppressive(e. g. , prednisone) and cytotoxic (e. g. , cyclophosphamide) therapies are commonly used without proven benefit.
• Thank you • Harrison and stepup