H HEMOSTAZA Szereg mechanizmw pozwalajcych na utrzymanie krwi


")


 dla czynników")











 plamice we wrodzonych")
 zmniejszone wytwarzanie płytek wrodzone: *")
")
- TROMBOCYTOPATIE: Wrodzone a) anomalie błony płytkowej: * zespół Bernarda-Souliera")


 I UZUPEŁNIAJĄCE W DIAGNOSTYCE ZABURZEŃ UKŁADU HEMOSTAZY NACZYNIOWE Czas krwawienia")





 Ca 2+")








 – gdy AT III - 25 –")




, koagulopatia ze zużycia, nabyta afibrynogenemia) -")




 PARAMETR PIERWOTNA FIBRYNOLIZA DIC FIBRYNOGEN PT TT")




- Slides: 56
H HEMOSTAZA – Szereg mechanizmów pozwalających na: · utrzymanie krwi w stanie płynnym w warunkach fizjologii · wykrzepianie, czyli utworzenie skrzepu w przypadku uszkodzenia naczynia · fibrynolizę, czyli rozpuszczenie skrzepu w celu udrożnienia światła naczynia Czynniki wpływające na hemostazę: płytki krwi skazy płytkowe • śródbłonek naczyń krwionośnych skazy naczyniowe · białka układu krzepnięcia (czynniki osoczowe) i fibrynolizy skazy osoczowe · Etapy hemostazy: pierwotna - wytworzenie miejscowego czopu płytkowego przy udziale płytek i pierwotna naczyń (obkurczenie naczyń adhezja i agregacja płytek) - czas trwania – 3 -5 min- metoda kontroli procesu – czas krwawienia
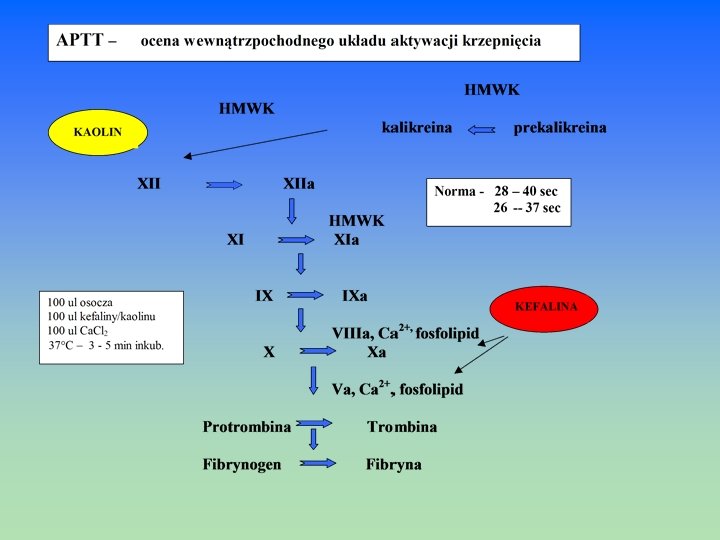
wtórna - wytworzenie skrzepu ostatecznego poprzez przekształcenie fibrynogenu wtórna w fibrynę na drodze wewnątrz- i zewnątrzpochodnej - czas trwania – 5 -10 min - metoda kontroli procesu – czas krzepnięcia zapoczątkowany przez układ wewnątrzpochodny układ zewnątrzpochodny * czas kaolinowo- kefalinowy = a. PTT = k-k * czas protrombinowy – PT metoda kontroli przekształcania fibrynogenu w fibrynę * czas trombinowy –T ostateczna - retrakcja wzmacniająca skrzep fibryny przy udziale trombasteniny ostateczna płytkowej - kończy się ok. 60 min od uszkodzenia naczynia - metoda kontroli procesu – retrakcja skrzepu osocza Fibrynoliza – rozpuszczenie skrzepu Fibrynoliza - czas trwania – 48 -72 godz. - metoda kontroli procesu – fibrynoliza skrzepu pełnej krwi lub osocza - fibrynoliza skrzepu euglobulin
I. ŚCIANA NACZYŃ KRWIONOŚNYCH Wewnętrzna warstwa – intima - poj. warstwa k. śródbłonka (endotelium) + podśródbłonkowa tkanka podskórna UTRZYMANIE PŁYNNOŚCI KRWI KRĄŻĄCEJ: * budowa - glikokaliks (glikozoaminoglikany (GAG)– 80 % -subst. heparynopodobne: i glikolipidy) - siarczan heparanu –SH - siarczan dermatanu -S *synteza subst. o właściwościach przeciwzakrzepowych : I. III. IV. V. VII. 1. TM – trombomodulina, wiążąca trombinę (T) TM -marker uszkodzenia nabłonka (wczesny marker DIC) kompleks TM-T Białko C (PC) aktywne białko C (APC) - inaktywacja cz. Va i VIIIa - aktywacja fibrynolizy Białko S (PS) – jako kofaktor białka C - tworzenie kompleksów z PF 3 i Ca 2. EPCR – śródbłonkowy rec. dla białka C (PC) 3. inhibitory krzepnięcia : AT III – antytrombina III t-PA – tkankowy aktywator plazminogenu u-PA – urokinazopodobny aktywator plazminogenu TFPI – inhibitor szlaku cz. tkankowego
4. czynniki naczynioworuchowe: Prostacyklina NO – tlenek azotu rozszerzanie naczyń krwionośnych (spadek ciśn. tętniczego) hamowanie aktywacji i agregacji płytek hamowanie adhezji płytek 5. ADP-aza – ADP adenozyna - hamowanie agregacji płytek Inaktywacja Va i VIIIa II. AKTYWACJA UKŁADU KRZEPNIĘCIA – HEMOSTAZA PIERWOTNA • Budowa – kolagen, fibronektyna, cz. v. W (aktywacja cz. XII, adhezja płytek) • Synteza subst. prozakrzepowych: Endotelina – skurcz naczyń PAF – cz. aktywujący płytki (uwalnianie serotoniny i TXA 2)
PŁYTKI KRWI = TROMBOCYTY – PLT • bezjądrzaste elementy morfotyczne krwi • powstają w szpiku w procesie trombopoezy z megakariocytów przy udziale trombopoetyny, Il-3, 11 • czas przeżycia płytek – 8 -12 dni (rozpad w ukł. s-ś śledziony i wątroby) wartości prawidłowe PLT = 150 – 400 x 103/ul (x 109/l) poziom hemostatyczny płytek (najmniejsza liczba płytek warunkująca czynność hemostatyczną płytek) PLT = 35 -40 x 103/ul poziom zagrażający życiu PLT poniżej 10 x 103/ul małopłytkowość PLT poniżej 100 x 103/ul nadpłytkowość PLT powyżej 600 x 103/ul
funkcja hemostatyczna uwarunkowana jest obecnością na powierzchni płytek • receptorów (glikoprotein-GP) dla czynników aktywacji receptorów (glikoprotein-GP np. Rec. Gp. Ib – rec dla cz. VW ( wiąże płytkę i kolagen-warunkując adhezję) Rec. Gp. IIb/IIIa – rec dla fbg (warunkuje agregację płytek) • różnych substacji zgromadzonych wewnątrz ziarnistości ziarnistości • swoiste białka płytek: PF 3, PF 4, -tromboglobulina • białka adhezyjne: cz. Von Willebranda, selektyna P, fibronektyna, trombospondyna, • czynniki krzepnięcia: fibrynogen, cz. V, cz. XI, HMWK • czynniki fibrynolizy: białko S, t. PA, PAJ-1 , inhibitor C 1 esterazy ziarnistości gęste jony Ca 2+ , ADP, ATP, TXA 2, serotonina lizosomy : peroksydazy: lizosomy kwaśne hydrolazy katalaza
FUNKCJE PŁYTEK KRWI
Adhezja płytek Gp I b + cz. VW + kolagen Aktywacja płytek - zmiana kształtu płytki - uwalnianie ziarnistości - udostępnienie FL do tworzenia kompleksu tenazy i protrombinazy Agregacja płytek Gp II b/IIIa + fbg Czop płytkowy
OSOCZOWE CZYNNIKI KRZEPNIĘCIA KRWI Synteza genetycznie uwarunkowana, geny kodujące syntezę większości czynników zlokalizowane są na chromosomach autosomalnych, z wyjątkiem genów kodujących cz. VIII i IX – w chromosomie płciowym X CZYNNIKI KONTAKTU Białka kofaktorowe , produkowane w wątrobie, niezbędne do aktywacji krzepnięcia w kontakcie z ujemnie naładowanymi powierzchniami: włókna kolagenu, kaolin, szkło Czynnik XII = cz. kontaktu = cz. Hagemana Czynnik XII Prekalikreina = Kalikreinogen = cz. Fletschera – proenzym kalikreiny, Prekalikreina - aktywator cz. XII i plazminogenu Wielkocząsteczkowy kininogen = cz. Fitzgeralda = HMWK – kofaktor aktywacji cz. XII, XI kininogen i kalikreinogenu Czynnik XI = cz. Rosenthala = cz. przeciwhemofilowy- C Czynnik XI CZYNNIKI ZESPOŁU PROTROMBINY Synteza w hepatocytach przy udziale witaminy K jako kofaktora. Czynnik X = cz. Stuarta-Prowera Czynnik X Czynnik IX = cz. Christmasa = cz. przeciwhemofilowy -B Czynnik IX Czynnik VII = Prokonwertyna = cz. stabilny Czynnik VII Czynnik II = Protrombina Czynnik II
CZYNNIKI WRAŻLIWE NA TROMBINĘ synteza w hepatocytach. Czynniki V i VIII są najbardziej labilnymi cz. i szybko ulegają degradacji w próbce krwi przechowywanej w temp. pokojowej lub w podgrzanym osoczu. Czynnik XIII = czynnik stabilizujący fibrynę, transglutaminaza osoczowa XIII Czynnik VIII = czynnik przeciwhemofilowy A, globulina antyhemofilowa VIII Czynnik V = proakceleryna Czynnik I = fibrynogen I Dodatkowe czynniki Czynnik III = tromboplastyna tkankowa = czynnik tkankowy (TF) = białko integralne III błon komórkowych m. in. fibroblastów, monocytów, makrofagów, rec. dla cz. VIIa 2+ Czynnik IV - jony Ca IV Cz. v. Willebranda – wyst. w osoczu i w płytkach krwi, ułatwia adhezję płytek tworząc v. Willebranda kompleks z cz. VIII, chroni go przed proteolityczną degradacją przez (APC) FL- Fosfolipidy błon komórkowych płytek – fosfatydyloseryna FL
Zespół tenazy Zespół protrombinazy
SCHEMAT HEMOSTAZY PIERWOTNEJ I OSTATECZNEJ 1. HEMOSTAZA NACZYNIOWA skurcz naczyń ( serotonina, TXA 2) 1. HEMOSTAZA NACZYNIOWA 2. HEMOSTAZA PŁYTKOWA czop płytkowy 3. HEMOSTAZA OSOCZOWA Fibrynogen Fibryna Fibrynogen (łań. Aα, Bβ, γ) Trombina odszczepia łańcuch Aα - Fibrynopeptyd A odszczepia łańcuch Bβ - Fibrynopeptyd B Monomery fibryny A + Monomery fibryny B labilny skrzep fibrynowy XIII a stabilizowana fibryna HEMOSTAZA OSTATECZNA
SCHEMAT UKŁADU FIBRYNOLITYCZNEGO FIBRYNOLIZA – rozpuszczenie złogów fibryny i utrzymanie drożności naczyń Plazminogen Układ wewnątrzpochodny Układ zewnątrzpochodny HWK-PK- XIIa -PA t-PA u-PA SK PAI-1 Plazmina cz. VIIIc cz. v. WF cz. XII 2 AP Fibrynogen Fibryna D-dimery, FDP Aktywacja hamowanie Skróty t-PA – tkankowy aktywator plazminogenu PAI – inhibitor aktywatora plazminogenu UK – urokinaza SK – streptokinaza 2 AP - 2 antyplazmina - 2 -makroglobulina FDP – produkty degradacji fibryny i fibrynogenu
plazmina Fibrynogen 2 monomeryczne fragmenty D tzw. FDP Fibrynogen 1 dimeryczny fragment E Funkcje FDP hamuje trombinę blokuje czynnik tkankowy • hamuje funkcję płytek (adhezję, agregację, r. uwalniania) Norma FDP 0 – 10 ug/ml FDP DIC z hiperfibrynolizą Pierwotna hiperfibrynoliza ! (po zabiegach na narządach z t-PA (płuca, macica, stercze) Zakrzepica żył głębokich Białaczki, nowotwory złośliwe Zawał m. sercowego Zator tętnicy płucnej Leczenie np. streptokinazą (wynik oznaczenia FDP obejmuje łącznie produkty degradacji fibrynogenu i fibryny) stabilizowana fibryna 2 fragmenty D połączone wiąz. krzyżowym przez cz. XIII 1 fragment E tzw. D- dimery Funkcje D-dimerów • Trawienie zakrzepu D-dimerów DIC , zakrzepica żył głębokich Norma D-dimerów do 200 ng/ml
NATURALNE INHIBITORY KRZEPNIĘCIA utrzymanie płynności krwi krążącej oraz zapobieganie nadmiernemu narastaniu czopu ostatecznego najważniejsze inhibitory: Antytrombina III – AT III Białko C (PC) Białko S (PS) jako kofaktor białka C Inhibitor zewnątrzpochodnego szlaku krzepnięcia –TFPI ANTYTROMBINA III - synteza w wątrobie, w mniejszym stopniu w śródbłonku naczyń oraz w megakariocytach - należy do rodziny serpin inaktywujących proteazy serynowe poprzez ich wiązanie w kompleks stechiometryczny w stos. 1: 1 - kofaktorem AT III jest heparyna, która tworząc z nią kompleks przyśpiesza reakcję inaktywacji cz. krzepnięcia 1000 x. Trombinę cz. XIIa AT III inaktywuje : cz. XI a cz. IX a cz. Xa, (ostatnio uważa się, że również VIIa)
BIAŁKO C - synteza formy nieaktywnej w wątrobie przy udziale witaminy K - aktywacja białka C uruchamiana jest przez pojawienie się trombiny we krwi trombina wiąże się w kompleks z trombomodulina inaktywacja cz. VIII a Białko C APC + PS inaktywacja cz. V a Niedobór białka C : DIC leczenie L-asparaginazą, doustnymi antykoagulantami TFPI - inhibitor zewnątrzpochodnego szlaku krzepnięcia - białko występujące w osoczu w formie związanej z Lp - w obecności cz. Xa wiąże i inaktywuje kompleks TF – VII a
SCHEMAT POŁĄCZEŃ UKŁADÓW KRZEPNIĘCIA, FIBRYNOLIZY, KININOGENEZY, KOMPLEMENTU I PŁYTEK
PODZIAŁ SKAZ KRWOTOCZNYCH NACZYNIOWE Wrodzone: wrodzona naczyniakowatość krwotoczna (ch. Rendu-Oslera) plamice we wrodzonych zaburzeniach tkanki łącznej np. zespół Marfana samoistna hemosyderoza płuc Nabyte: zespół Schönleina-Henocha plamice w przebiegu zakażeń plamice w przebiegu amyloidozy plamica starcza, ortostatyczna, mechaniczna, polekowe, z zapaleniem naczyń włosowatych, szkorbut - rozpoznanie : przedmiotowe bad. lekarskie + wywiad rodzinny wyniki testów z zakresu krzepnięcia krwi i hemostazy - norma
PŁYTKOWE Zmiany ilościowe: A. Małopłytkowości - Trombocytopenie a) zmniejszone wytwarzanie płytek wrodzone: * wrodzona hipoplazja szpiku – zespół Fanconiego nabyte : * nk aplastyczna * aplazja megakariocytowa * nacieczenie szpiku kostnego (ALL, chłoniaki, gruźlica) * zwłóknienie szpiku * leki mielosupresyjne, zw. chemiczne, promienie jonizujące * nk megaloblastyczne, z niedoboru żelaza, nocna napadowa hemoglobinuria * zakażenia wirusowe, niewydolność nerek b) nadmierne niszczenie płytek wrodzone: * samoistna autoimmunologiczna plamica małopłytkowa nabyte: immunologiczne: * małopłytkowość poprzetoczeniowa, polekowa * autoimmunologiczna nk hemolityczna * toczeń rumieniowaty układowy * wstrząs anafilaktyczny
nieimmunologiczne: * DIC * zakażenia * zespół hemolityczno- mocznicowy * zakrzepowa plamica małopłytkowa c) nieprawidłowe rozmieszczenie płytek w ustroju * hipersplenizm d) utrata płytek: * krążenie pozaustrojowe, krwotoki B. Nadpłytkowości pierwotne: * samoistna, * zespóły mieloproliferacyjne wtórne: * stany zapalne (gruźlica, sarkoidoza, RKZ, wrzodziejące zap. jelita grubego) * ch. nowotworowe * po splenektomii i innych zabiegach pooperacyjnych * po krwotokach * w niedoborze żelaza * polekowa ( np. winkrystyna) * powysiłkowa (trwająca ok. 15 -30 min)
Zmiany jakościowe (zaburzona czynność płytek)- TROMBOCYTOPATIE: Wrodzone a) anomalie błony płytkowej: * zespół Bernarda-Souliera (defekt kompleksu GP Ib/IX/V- płytkowy rec. dla cz. v. W zaburzona adhezja płytek krwi * trombastenia Glanzmanna ( defekt kompleksu GP IIb/IIIa – rec. dla fbg) zaburzona agregacja płytek krwi * defekt receptora kolagenu ( defekt GP Ia/IIa) - łagodna skaza krwotoczna * zespół Scotta (zaburzenia prokoagulacyjnej czynności płytek) b) zaburzenia sekrecji ziarnistości płytkowych B. Nabyte : * wpływ leków * ch. krwi (ALL, MDS, zespoły mieloproliferacyjne), * DIC * inne choroby np. mocznica, krążenie pozaustrojowe, przewlekłe ch. wątroby
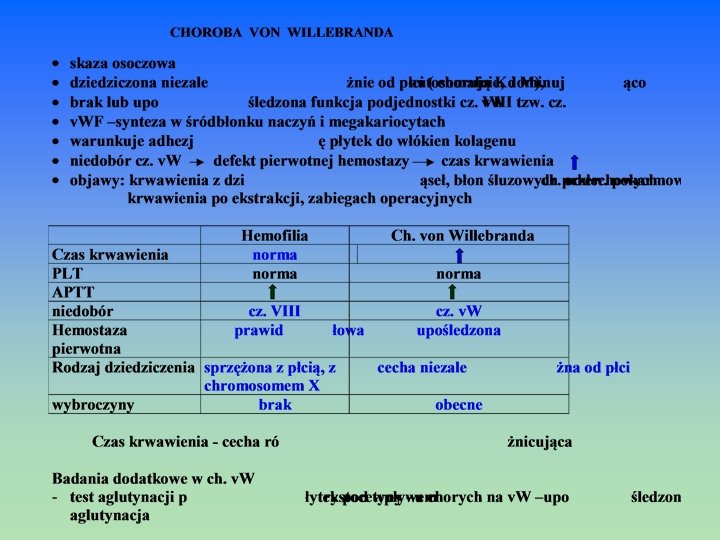
OSOCZOWE Wrodzone: Wrodzone * Ch. von Willebranda * Hemofilia A *Hemofilia B * A-, hipo-, dysfibrynogenemia * Niedobór pojedynczego cz. II, V, VII, X, XII, XIII (występują rzadko) Nabyte: a) niedobór witaminy K upośledzenie wytwarzania wit. K np. * ch. krwotoczna noworodków upośledzenie wchłaniania wit. K *kamica, nowotwór, zespół złego wchłaniania upośledzone wykorzystanie wit. K * doustne antykoagulanty-pochodne dihydroksykumaryny b) zaburzenia krzepnięcia z powodu chorób wątroby
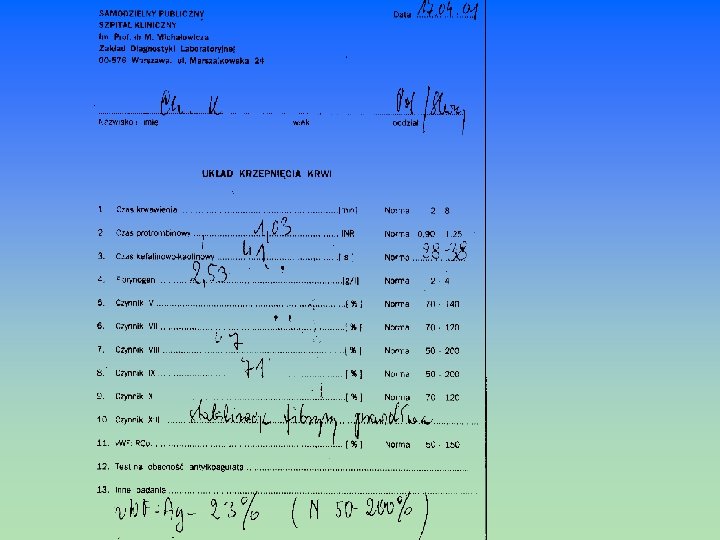
ZASADY POBIERANIA KRWI DO BADAŃ UKŁADU KRZEPNIĘCIA RANO, NA CZCZO ( lub lekki posiłek beztłuszczowy) W WARUNKACH SPOKOJU, BEZ STRESU KREW ŻYLNA NA 3, 2% CYTRYNIAN SODU (1 cz. cytrynianu - 9 cz. krwi ) BEZ STAZY lub UCISK ok. 30 sek OSTRE, DOŚĆ GRUBE IGŁY KREW PO ODRZUCENIU PIERWSZYCH 2 -3 ML PLASTIKOWE PROBÓWKI BARDZO DELIKATNE MIESZANIE KRWI TUŻ PO POBRANIU (3 -4 krotne odwrócenie probówki do góry dnem - nie wolno spienić! ) NIEZWŁOCZNIE ODWIROWAĆ PRÓBKĘ KRWI (10 min – 3 tys. obrotów/min) BADANIA WYKONAĆ W CIĄGU MAX 2 GODZIN OD POBRANIA
BADANIA PRZESIEWOWE (PODSTAWOWE) I UZUPEŁNIAJĄCE W DIAGNOSTYCE ZABURZEŃ UKŁADU HEMOSTAZY NACZYNIOWE Czas krwawienia metodą Ivy Testy specjalistyczne – test oporności kapilarowej - biopsja skóry PŁYTKOWE Czas krwawienia - BT Liczba płytek krwi –PLT Rozmaz krwi obwodowej Badania czynnościowe: Adhezja i agregacja płytek Reakcja uwalniania Ocena zawartości ziarnistości płytek Czas zużycia protrombiny OSOCZOWE Czas kaolinowo-kefalinowy – a. PTT – (k-k) -czas częściowej tromboplastyny po aktywacji Czas protrombinowy – PT Czas trombinowy – TT Czas rekalcynacji – CT Fibrynogen Poszczególne czynniki krzepnięcia: VIII, VII, XIII Krążące antykoagulanty Inhibitory krzepnięcia: Antytrombina III –AT III Białko C, białko S
FIBRYNOLITYCZNE Czas lizy euglobulin Czas trombinowy FDP, D-dimery Plazminogen Aktywatory fibrynolizy: Tkankowy aktywator plazminogenu – t-PA Urokinaza – u-PA Inhibitory fibrynolizy: Inhibitor aktywatora plazminogenu – PAI-1 2 antyplazmina - 2 AP
CZAS KRWAWIENIA Czas od chwili uszkodzenia naczynia do samoistnego ustania krwawienia • jest jednoznaczny z czasem trwania hemostazy pierwotnej • jest miarą: czasu utworzenia czopu płytkowego skurczu naczyń adhezji i agregacji płytek do śródbłonka naczyń • próba czynnościowa płytek krwi zależna częściowo od stanu naczyń, nie zależy od czynników krzepnięcia • metody oznaczania : Metoda Duke’a Nakłucie skóry opuszki palca na głębokość 2 -3 mm i pomiar czasu do momentu zaprzestania wypływu krwi Norma - do 5 min
Metoda Ivy z modyfikacją Mielke – met. zalecana Standaryzacja pomiaru – pomiar czasu wypływu krwi od momentu dokonania nacięcia na skórze przedramienia o dł. 10 mm i głębokości ok. 2, 5 mm (zestawy np. Simplate) przy ciśn. 40 mm. Hg (ciśn. nadmuchane po założeniu opaski ciśnieniomierza) do braku śladu krwi na przykładanej do nacięcia bibule filtracyjnej. Norma – 2 – 10 min (do 8 min) CZAS KRWAWIENIA • małopłytkowość • trombastenia Glanzmana (wrodzone zaburzenia agregacji płytek) • ch. von Willebranda • niektóre trombocytopatie • skazy krwotoczne z hipofibrynogenemią • po aspirynie
CZAS REKALCYNACJI OSOCZA - CK czas krzepnięcia po dodaniu do osocza nadmiaru jonów Ca 2+ w probówce szklanej ocenia aktywność układu wewnątrzpochodnego Norma CT: 100 – 210 sek próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś czynnika poniżej 3 – 5% normy ( skazy krwotoczne wyst. już przy wartościach 10 – 20 % normy) CZAS KRZEPNIĘCIA WG. METODY LEE – WHITE’A (CZAS L-W) – WHITE’A (CZAS L-W) pomiar czasu od pobrania pełnej krwi do jej skrzepnięcia w szklanej probówce w temp. 37 C określa sprawność układu wewnątrzpochodnego (próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś czynnika poniżej 1 – 3 % normy) Norma czasu L-W : 8 – 12 min Czas krzepnięcia · niedobór cz. układu wewnątrzpochodnego (XII, XI, IX, VIII) · niedobór cz. drogi wspólnej ( X, V, II, I) · obecność inhibitorów czynników krzepnięcia np. podczas leczenia heparyną
PT – ocena zewnątrzpochodnego układu krzepnięcia Czynnik VII a, cz. tkankowy (TF) Ca 2+ Czynnik X czynnik X a tromboplastyna czynnik Va, Ca 2+, FL protrombina fibrynogen fibryna 100 ul osocza 100 ul tromboplastyny 100 ul Ca. Cl 2 SPOSOBY WYRAŻANIA CZASU PROTROMBINOWEGO CZAS PROTROMBINOWY WSKAŹNIK PROTROMBINOWY Czas (sekundy) PT prawidłowy x 100 PT pacjenta WSPÓŁCZYNNIK PROTROMBINOWY - R PT pacjenta PT prawidłowy INR – międzynarodowy współczynnik znormalizowany R ISI – międzynarodowy indeks czułości 11 – 13 sek 12 – 16 sek 80 – 120 % 0, 85 – 1, 15 0, 9 – 1, 25
TT – ocena przejścia fibrynogenu w fibrynę – ocena drogi wspólnej Trombina Norma TT – ok. 15 sek TT zależy od: stęż. Fbg, trombiny, aktywności AT III, prawidłowej stabilizacji fibryny
TT Hipofibrynogenemia np. w marskości wątroby, DIC i afibrynogenemii 0 g/l Dysfibrynogenemia Obecność inhibitorów trombiny np. leczenie heparyną Obecność inhibitorów polimeryzacji fibryny np. FDP Inne: mocznica, gammapatia monoklonalna TT Nadkrzepliwość TT wykonać Czas reptylazowy pomiar czasu krzepnięcia osocza po aktywacji reptylazą (wyciąg z jadu węża) - enzym o działaniu podobnym do trombiny, ale niezależny od heparyny norma – ok. 18 – 20 sek TT + t. reptylazowy obecność FDP (hamują polimeryzację fibryny) TT + N t. reptylazowy obecność heparyny (hamuje trombinę (nie hamuje reptylazy)
FIBRYNOGEN - FBG • Glikoproteina syntetyzowana w wątrobie • niezbędna w procesie krzepnięcia do tworzenia czopu płytkowego (warunkuje agregację płytek) i ostatecznego (tworzenie siatki fibrynowej) • Białko ostrej fazy, którego stęż. rośnie w początkowym okresie infekcji (wpływ na wzrost OB) • niezależny czynnik ryzyka choroby wieńcowej, zawału m. sercowego, udaru mózgu (bierze udział w transporcie chol i tworzeniu k. piankowatych, powoduje rozrost mięśni gładkich – zw. miażdżycorodny) wzrost między 3 -5 dniem w zawale, stabilizacja w ciągu 20 dni im wyższy fbg, tym gorsze rokowanie w zawale Norma - 2, 0 – 5, 0 g/l 200 -500 mg/dl Metody oznaczania fbg: • metoda chronometryczna Claussa (ścisła korelacja między trombinowym czasem krzepnięcia i stężeniem fibrynogenu w osoczu) • metoda kolorymetryczna z odczynnikiem Folina i Ciocalteu
Fibrynogen • DIC • pierwotna hiperfibrynoliza • wrodzony brak lub niedobór fbg • marskość wątroby i inne uszkodzenia czynności wątroby • ch. hematologiczne (AL, nk aplastyczna) • podczas leczenia streptokinazą Fibrynogen hiperfibrynogenemia przemijająca: Hiperfibrynogenemia dłużej trwająca • III trymestr ciąży i okres okołoporodowy nowotwory złośliwe • po zabiegach operacyjnych przewlekłe stany zapalne • zakrzepica kolagenozy • urazy i ostre stany zapalne, zwłaszcza zapalenie płuc zespół nerczycowy • zawał serc
FIBRYNOLIZA SKRZEPU EUGLOBULIN OSOCZA - próba ogólna układu fibrynolitycznego - czas upłynnienia skrzepu euglobulin osocza wytrąconych pod wpływem roztworu o niskiej sile jonowej, p. H-5 i temp. 4 C, a następnie rozpuszczonych w buforze i wykrzepionych Ca. Cl 2 - mierzy się czas od momentu powstania skrzepu do chwili rozpuszczenia w temp. 37 C - euglobulinowa frakcja białkowa zawiera : plazminogen plazmina aktywatory plazminogenu fibrynogen cz. krzepnięcia: m. in. VIII, XIII - brak inhibitorów plazminogenu np. antyplazmin (które pozostają w supernatancie) Norma - 120 – 240 min (2 – 4 godz. )
czas lizy euglobulin III trymestr ciąży okres pooperacyjny - choroby zatorowo-zakrzepowe - zawał serca i miażdżyca - cukrzyca - nadciśnienie czas lizy euglobulin skazy krwotoczne przebiegające z hiperfibrynolizą - zaawansowana choroba nowotworowa - marskość wątroby - ostre stany septyczne - po przetoczeniu krwi obcej grupowo przy znacznym skróceniu czasu lizy euglobulin przed operacją podaje się leki hamujące aktywność t-PA
ANTYTROMBINA III – AT III - badanie aktywności czynnej trombiny pozostałej po inaktywacji przez kompleks AT III – heparyna i trawiącej substrat chromogenny - ilość wolnej trombiny jest odwrotnie proporcjonalna do aktywności AT III Norma dla AT III Stęż. ATIII w osoczu – 22 -40 mg/dl Aktywność AT III 1 dzień ż. – 39 - 87 % 1 m. ż. - 48 – 108 % 3 m. ż. - 73 - 120 % pozostali - 75 – 125 % aktywności AT III ( zaburzone krzepnięcie) WZW Leczenie steroidami anabolicznymi Chorzy po przeszczepie nerki Leczenie doustnymi antykoagulantami
Aktywności AT III (tendencja do nadkrzepliwości) – gdy AT III - 25 – 50 % normy • wrodzony niedobór AT III (2 x częstszy od hemofilii) • nabyty niedobór: a) zwiększone zużycie * DIC * stany zakrzepowo-zatorowe * zawał serca * stany pooperacyjne , * wstrząs endoseptyczny, * masywne oparzenia * używanie doustnych środków antykoncepcyjnych b) zmniejszona synteza - * ch. wątroby c) nadmierne wydalanie - * enteropatia jelitowa z utratą białek - * ch. nerek : zespół nerczycowy, kłębkowe zapalenie nerek Leczenie preparatem AT III do uzyskania aktywności bezpiecznej – 80 % wskazania: - terapia substytucyjna we wrodzonych niedoborach AT III - u chorych z DIC - u chorych ze wstrząsem septycznym monitorowanie dawkowania należy prowadzić, oznaczając aktywność a nie stężenie AT III ( bo szybciej wraca do normy) co 4 -6 godz
ALGORYTM CZASÓW
APTT PT - N, TT - N , BT - N niedobór cz. XII, XI, IX, VIII (hemofilia lub ch. v. W), PK obecność krążącego antykoagulantu próba korekcji APTT prawidłowa brak korekcji niedobór czynników krążący antykoagulant (anty-VIII lub LA) oznaczyć poziom poj. czynników miano APTT PT TT – N, BT -N niedobór cz. II, V, X, VII (choroby wątroby) złożony niedobór cz. zależnych od witaminy K (znaczny) zaburzenia po masywnych przetoczeniach próba korekcji APTT oznaczyć poziom cz. II, V, VII, X APTT PT TT BT – N hypo- i dysfibrynogenemie DIC ch. wątroby oznaczyć stęż. fibrynogenu aktywacja fibrynolizy FDP wpływ heparyny czas reptylazowy APTT – N PT TT – N, BT – N niedobór cz. VII rozpoczęcie leczenia antykoagulantami doustnymi APTT- N, PT – N, TT- N, BT ch. von Willebranda APTT – N, PT – N, TT- N, BT zaburzenia płytkowo-naczyniowe
TROMBOFILIA – NADKRZEPLIWOŚĆ Wrodzona lub nabyta skłonność do zakrzepów przyczyny zaburzeń nabytych: obecność p/c antyfosfolipidowych (Lupus antykoagulant- LA) nadpłytkowość czerwienica prawdziwa zwiększona aktywność inhibitorów fibrynolizy przyczyny wrodzonej trombofilii ( wyst. poniżej 40 r. ż. , bez czynników ryzyka): oporność na aktywne białko C (APC-R) – 12 – 64 % częstość występowania niedobór białka C - 5 – 8 % niedobór białka S - 5 – 8 % niedobór AT III - 2 – 4 % APC-R zaburzenie związane z mutacją punktową w obrębie cz. V typu Leiden, zmieniony cz. V ma aktywność prokoagulacyjną, natomiast nie ulega proteolizie pod wpływem APC
BADANIE FIZYKALNE
ZESPÓŁ WYKRZEPIANIA ŚRÓDNACZYNIOWEGO - DIC (disseminated intravascular coagulation), koagulopatia ze zużycia, nabyta afibrynogenemia) - nabyty zespół zaburzeń krzepnięcia krwi , wtórny do podstawowej jednostki chorobowej - uogólniona wewnątrznaczyniowa aktywacja krzepnięcia prowadząca do wytworzenia złogów fibryny z towarzyszącą lub zahamowaną wtórną aktywacją fibrynolizy Najczęstsze stany kliniczne prowadzące do DIC: • zakażenia: bakteryjne (posocznica), wirusowe, grzybicze, riketsjozy • powikłania ciąży i porodu • nowotwory: rak gruczołu krokowego, trzustki, płuc, okrężnicy • ch. ukł. krwiotwórczego: AML-M 3, przełomy hemolityczne, hemoliza wewnatrznaczyniowa po toczeniu nieprawidłowej krwi • choroby serca i naczyń krwionośnych: naczyniaki, zator tętnicy płucnej • rozległe uszkodzenia tkanek: oparzenia, zabiegi operacyjne • choroby wątroby • inne choroby: OZT, rozległa zakrzepica żył głębokich, odczyny alergiczne na leki
Mechanizmy odpowiedzialne za patogenezę DIC choroba leżąca u podłoża DIC (sepsa, uraz, itd. ) cytokiny pobudzenie syntezy cz. III zużycie inh. krzepnięcia zwiększenie syntezy PAI-1 aktywacja krzepnięcia osłabienie fibrynolizy poprzez cz. VII tworzenie złogów fibryny zmniejszone usuwanie fibryny zużycie cz. krzepnięcia tworzenie zakrzepów w i płytek mikrokrążeniu aktywacja fibrynolizy tworzenie FDP skaza krwotoczna uszkodzenie narządów
OBJAWY KLINICZNE DIC krwawienia z miejsc wkłuć do żyły - krwawienia z dróg rodnych, moczowych, z przewodu pokarmowego - wybroczyny na skórze, błonach śluzowych - krwotoki z ran operacyjnych i pourazowych + zakrzepica i uszkodzenie serca, płuc, nerek, wątroby, OUN (ostra postać DIC) + ogólne objawy : gorączka, kwasica, białkomocz, hipoksja BADANIA PODSTAWOWE W DIC PT PLT APTT FBG TT FDP D-dimery + testy parakoagulacji rozmaz krwi obwodowej- schizocyty 50%, duże płytki, wzrost WBC i Ret, nk hemolityczna mikroangiopatyczna
BADANIA UZUPEŁNIAJĄCE AT III cz. V, VIII białko C DIAGNOSTYKA pre-DIC Testy wczesnego ostrzeżenia – 7 dni przed wystąpieniem objawów DIC Markery aktywacji hemostazy SF – rozpuszczalna fibryna TAT – kompleks ATIII- trombina PAP - kompleks plazmina-antyplazmina D- dimery Markery uszkodzenia śródbłonka TM – trombomodulina ( wzrost na 1 dzień przed objawami) TPA – tkankowy aktywator plazminogenu PAI - inhibitor aktywatora plazminogenu v. WF – cz. von Willebranda
LABORATORYJNE KRYTERIA DIAGNOSTYCZNE DIC I. Testy wykrywające aktywację układu krzepnięcia F 1+2 – fragment protrombiny, powstaje pod wpływem cz. Xa na cz. II (czuły marker aktywacji ukł. wewnątrzpochodnego) TAT – kompleks ATIII- trombina FA i FB - fibrynopeptyd A i B II. Testy wykrywające aktywację układu fibrynolizy D- dimery FDP Plazmina PAP – kompleks plazmina – antyplazmina III. Testy wykrywające zużycie inhibitorów krzepnięcia i fibrynolizy AT III TAT α 2 antyplazmina PAP białko C i S IV. Testy wykrywające uszkodzenie narządów LDH p. H - kwasica Kreatynina stęż. prężności tlenu - hypoksja Do spełnienia kryteriów laboratoryjnych rozpoznania DIC potrzeba jednego nieprawidłowego wyniku z każdej grupy testów I, II i III oraz dwóch z grupy IV.
Różnicowanie pierwotnej i wtórnie zaktywowanej fibrynolizy (DIC) PARAMETR PIERWOTNA FIBRYNOLIZA DIC FIBRYNOGEN PT TT APTT PLT NORMA CZAS LIZY EUGLOBULIN MONOMERY FIBRYNY FDP, D-DIMERY BRAK OBECNE CZ. V, VIII AT III NORMA
HHEMOFILIE - skaza krwotoczna osoczowa wwrodzone zaburzenie krzepnięcia krwi związane z chromosomem X (dziedziczenie recesywne sprzężone z płcią - chorują chłopcy, kobiety są nosicielami, chorują przy spadku poziomu czynnika VIII poniżej 40 %): hemofilia A – wrodzony niedobór cz. VIII hemofilia B – wrodzony niedobór cz. IX hemofilia C – wrodzony niedobór cz. XI HEMOFILIA A - wrodzony niedobór cz. VIII, stopień ciężkości choroby zależy od aktywności cz. VIII - Postać hemofilii Aktywność cz. VIII w surowicy Objawy ciężka poniżej 1 % średnio ciężka 1 – 5 % łagodna 5 – 15 % częste nawracające krwawienia do stawów (zniekształcenia), do mięśni i narządów wewnętrznych krwawienia samoistne (rzadko) Ciężkie krwawienia po ekstrakcji zębów, zabiegach chirurgicznych, krwiaki pourazowe Krwawienia po zabiegach, urazach subhemofilia 15 – 30 % bezobjawowo
Rozpoznanie hemofilii - objawy kliniczne + wywiad rodzinny - badania : APTT - ! - sprawdzić aktywność cz. VIII PLT - N prawidłowa hemostaza pierwotna (brak wybroczyn na skórze) Czas krwawienia – N PT - N Podanie dawki cz. VIII Dawka = m. ciała (kg) x pożądany wzrost aktywności (w %) x 0, 5 Podanie preparatu – sprawdzenie APTT brak skrócenia Obecność autoprzeciwciał przeciw cz. VIII lub krążącego antykoagulanta
SCHEMAT DZIAŁANIA HEPARYNY I DOUSTNYCH ANTYKOAGULANTÓW Heparyna Doustne antykoagulanty UFH – heparyna wielkocząsteczkowa, niefrakcjonowana Heparyny stosowane są u pacjentów: z żylną i tętniczą zakrzepicą w żylnej profilaktyce ch. zatorowo-zakrzepowej Obie heparyny działają pośrednio poprzez wpływ na AT III (tworzenie kompleksu) LMWH UFH hamowanie cz. Xa -hamowanie cz. Xa i trombiny - brak konieczności monitorowania leczenia -monitorowanie leczenia UFH poprzez APTT
pobieranie krwi: bezpośrednio przed podaniem heparyny po 6 godz – w przypadku wlewu ciągłego dożylnego po 4 -5 godz - przy podaniu podskórnym terapeutyczne stęż. heparyny – 1, 5 - 2, 5 x wzrost APTT w porównaniu z APTT przed leczeniem Objawy uboczne UFH : małopłytkowość na poczatku leczenia, utajony wrzód żołądka lub dwunastnicy, skaza krwotoczna, osteoporoza (po okresie 3 miesięcy) (hirudyna -wyciąg ze ślinianek pijawek lekarskich -działa bezpośrednio na AT III ANTYKOAGULANTY DOUSTNE - hamują cykl przemian wit K hamowanie produkcji cz. wit. K zależnych (II, VII, IX, X) • hamują produkcję białka C i S • monitorowanie leczenia poprzez pomiar PT terapeutyczne stęż. leku pobieranie krwi: bezpośrednio przed podaniem INR – 2, 0 – 3, 0 oraz w 3, 5, 7 dniu leczenia INR – 2, 5 -3, 5 (w ciężkich przypadkach) po ustaleniu dawki - co tydzień w pierwszym m-cu leczenia co 2 -3 tyg. w II i III m-c co 4 -6 tyg w okresie póżniejszym