ENFERMEDADES DE LAS MOTONEURONAS T Sevilla Hospital La


 y corticobulbar (TCB)")
")

 Infecciones (mielitis retrovirales, HIV,")


, sexual, parenteral. • Inicio después de")


 Pérdida")




 •")












 Excitotoxinas Estrés oxidativo Disfuncion neurofilamentos")
 20% de ELA familiar, 1%")


 Reposo, b) protrusión A B")


 evidencia de afectación MN inferior (MNI),")
 evidencia electrofiológica")
 Siringomielia, siringobulbia (RM) Mielopatía cervical espondiloartrósica (RM)")
 • tratamiento sintomático (dietético,")

- Slides: 46
ENFERMEDADES DE LAS MOTONEURONAS T. Sevilla Hospital La Fe
Enfermedades de las motoneuronas • Enfermedad de la MN superior Esclerosis Lateral Primaria Paraplejía espástica hereditaria Parapesia espástica tropical (HTLV-1) Adrenomieloneuropatía • Enfermedad de la MN inferior Poliomielitis aguda Atrofia bulbar progr. infancia Síndrome postpolio Enf. Kennedy/atrofia bulboespina Amiotrofia focal benigna Atrofia ms. progresiva Atrofia ms espinal inf/juvenil (AME) AME adulto • Enfermedad de la MN superior e inferior Esclerosis Lateral Amiotrófica (ELA)
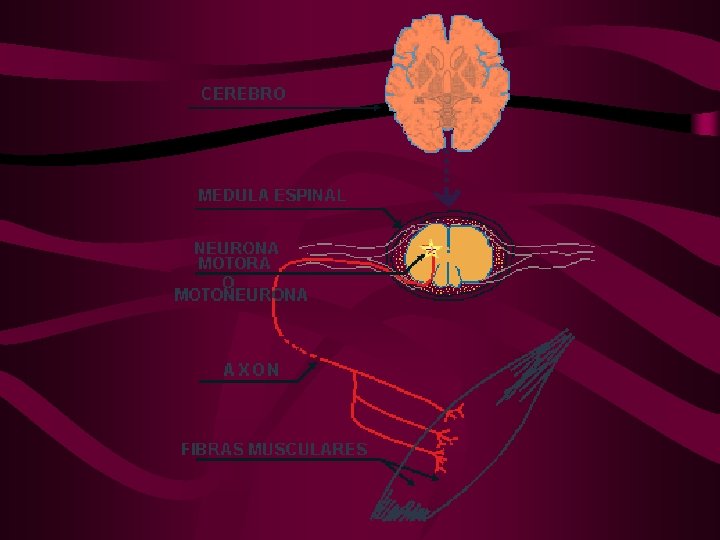
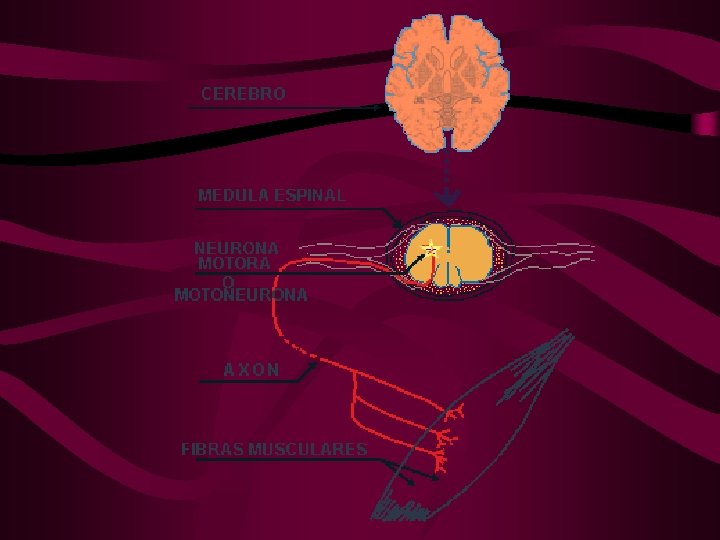
Enfermedad MNS: anatomía • Cels. están en cortex: tractos corticoespinal (TCE) y corticobulbar (TCB) • MNS está rostral a MNI controla su actividad. • TCB proyecta a núcleos de V, VII, IX, X y XII. • TCE lateral (tracto piramidal) y proyecta a las IN y MN del asta anterior de ME, controlan los mov. extremidades. • TCE anterior no se cruza proyecta a las IN y MN que controlan mov. axiales. • Los ax. TCE proveen actividad excitatoria alfa glutamatérgica a las alfa MN
Síntomas y signos MNS • • • Pérdida destreza (lentitud al ejecutar mov. ) Pérdida fuerza muscular (debilidad) Espasticidad ( tensión ms al estiramiento) Hiperreflexia y reflejos patológicos (Babinski) Parálisis pseudobulbar (risa y llanto espontáneo e inmotivado) • Espasmos flexores (movimientos bruscos piernas)
ESCLEROSIS LATERAL PRIMARIA: Clínica • Incidencia: 2 -4% ELA. • No signos de afectación MNI • Inicio aprox. 50 a. paraparesia espástica que asciende a MS y eventualmente ms. bulbar • Signos clínicos de afectación MNS • Curso lento, tras años puede aparecer atrofia • No alteración de la sensibilidad
ELP: diagnóstico dif. y trto • • • Tumores (RM) Infecciones (mielitis retrovirales, HIV, HTLV-1), Enf. desmielinizantes (LCR, RM) Adrenomieloneuropatía (Ac. grasos c larga ) Mielopatía cervical espondilótica (RM) Degeneración combinada subaguda (B 12) Encefalomielitis paraneoplásica (Ac anti Hu) Paraparesia espástica hereditaria (genética) Mielopatía tiroidea e hiperparatiroidea (Ca y P) Trto: antiespásticos como Baclofén y tizanidina
PARAPARESIA ESPASTICA HEREDITARIA • Espasticidad de EI, variable grado debilidad • Formas puras y complicadas (asociadas a NO, sordera, n. periférica, alt. extrapiramidales etc. ) • • Prevalencia: 0. 5 -11. 9/100. 000 Herencia: mayoría AD, tb. AR y X-L Genética: 40 -50%, espastina (>30 genes/loci). Diagnóstico: Hª familiar, si no hay, excluir otras causas (ELP, etc)
MIELOPATIA ASOCIADA A HTLV-1: PARAPESIA ESPASTICA TROPICAL • HTLV-1 (Human T-cell lymphotrophic virus, Type I) es un retrovirus que causa mielopatía progresiva de EI. • Áreas: Japón, Caribe, África tropical, áreas de centro y Sudamérica. • Grupos de riesgo: Adictos drogas vía parenteral, inmigrantes de países de alto riesgo, frecuente asociado a coinfec. HTLV-II y HIV • Mielopatía en 1% de los infectados, la mayoría asintomáticos.
MIELOPATIA ASOCIADA A HTLV-1 • Transmisión: Vertical (madre-hijo), sexual, parenteral. • Inicio después de los 30 a. con paraparesia espástica, neuropatía sensit. dolorosa, alt. esfínteres y a veces neuritis óptica. • LCR: proteínas altas, pleocitosis • Diagnóstico: serología positiva • Asociada a HTLV-II: evolución más rápida
ADRENOMIELONEUROPATIA • Adrenoleucodistrofia: niños 4 -8 años con insuf. adrenal, demencia, cuadriparesia, ceguera, sordera y crisis epilépticas. • Adrenomieloneuropatía: paraparesia espástica y neuropatía en jóvenes o adultos • Herencia: X-L, las mujeres portadoras pueden tener paraparesia leve. • Dx: acúmulo de ac. grasos de cadena muy larga en plasma, eritrocitos o fibroblastos
ENFERMEDADES DE LAS MNI: Neuroanatomía • Interneuronas: la mayoría cels. del aa ME • Reciben el control motor supranuclear excitatorio e inhibitorio de los tractos motores CE, CB, sistema límbico y NP. • Esta red media mov. reflejos y voluntarios • Todas estas vías IN convergen en MNI que inervan los ms. esqueléticos • MNI: en TE y ME, envían axones a fibras ms
Síntomas y signos afectación MNI • • • Pérdida de fuerza muscular (debilidad) Pérdida de masa muscular (atrofia) Hipotonía Hipo o arreflexia Fasciculaciones: contrac. ms. espontáneas, por si solas no indican enfermedad. • Calambres: mov. abruptos, involuntarios y dolorosos del músculo. Mejoran con masaje
MNI: laboratorio • Examen electrodiagnóstico: las fibras que han perdido inervación descargan actividad espontánea eléctrica (fibrilac. , ondas+) • Biopsia muscular: atrofia de grupos de fibras musculares junto a fascículos indemnes.
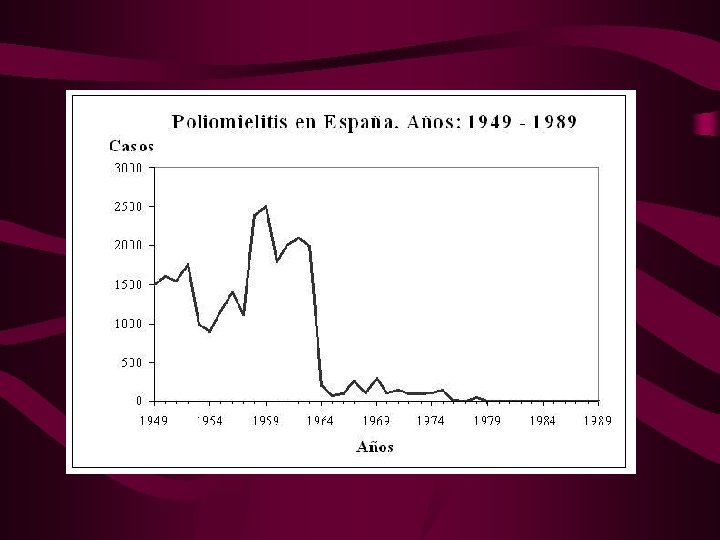
POLIOMIELITIS AGUDA: síntomas • Etiología : poliovirus, trans. feco-oral, entra en tej. linfoide, sangre y SNC. • Incubación, 3 -6 días • Viremia 2 -3 días: 90% asintomáticos, 10% síndr. gripal con fiebre, tos diarrea, etc. • < 1% síndrome paralítico agudo (mialgia, fasciculaciones, parálisis focal fulminante y asimétrica) • Afecta cualquier ms (> extrem, < bulbares)
POLIOMIELITIS : signos • • Debilidad Reflejos deprimidos o ausentes Hipotonía Hipo o arreflexia Fasciculaciones Atrofia Tras la 1ª semana se inicia la mejoría, 2/3 quedan con algún daño funcional.
POLIOMIELITIS: diagnóstico • Electrofisiología: signos denervación, fasc. • LCR: pleocitosis, 1º polim. después linfos • Diagnóstico específico: identificando Ac Ig. M en LCR contra poliovirus • Documentar título de Ac en suero contra el poliovirus (más de 4 veces)
POLIOMIELITIS: D. Diferencial y prevención • Encefalopatía por el v. del Nilo (serología) • S. Guillain-Barré (LCR no cels, EMG desmielinizante) • M gravis, neuralgia amiotrófica, parálisis periódica, polineuropatía por garrapatas. • Tratamiento: medidas de soporte y RHB • Prevención: vacuna trivalente Sabin que contiene los tres serotipos vivos atenuados. • Si se viaja a lugares de prevalencia: 1 dosis
SINDROME POSTPOLIO • Pacientes que han padecido la polio y han tenido un curso estable de al menos 10 a. • Clínica: puede aparecer dolor, fatiga, calambres, disartria, disfagia, intolerancia al frío y aumento de la atrofia. • Diagnóstico: por exclusión • Tratamiento: medidas de soporte, evitar actividades que causen fatiga.
• Etiopatogenia: una n. Motora que, en un principio, inervaba 1000 cels ms, puede acabar inervando 5. 000 -10. 000 células, creando una unidad motora (UM) gigante. Estas adaptaciones no son estáticas ni permanentes. Tras la recuperación de la PM se asiste a un proceso de remodelación de las UM, con degeneración de los extremos de los axones (pérdida de viejas dendritas) y reinervación contemporánea (aparición de nuevas dendritas). Ese proceso hace que las UM mantengan la función muscular en un estado de equilibrio dinámico. Cuando se altera ese equilibrio, reaparece la debilidad y, ocasionalmente, la parálisis.
NEUROPATIA MOTORA MULTIFOCAL Se ha dado en el tema de neuropatías
AMIOTROFIA FOCAL BENIGNA • Inicio adolescencia, a veces hasta 4ª-5ª dec. • Afecta preferentemente a sexo masculino, se describió en Japón e India (Hirayama) • Atrofia idiopática progresiva de mano/antebrazo • Hipo o arreflexia en segmento afecto • No signos de MNS • Dx: EMG, RM cervical • Dx dif: ELA, NMM, radiculopatías, N. amiotro, siringomielia, tumor de las vainas nerviosas
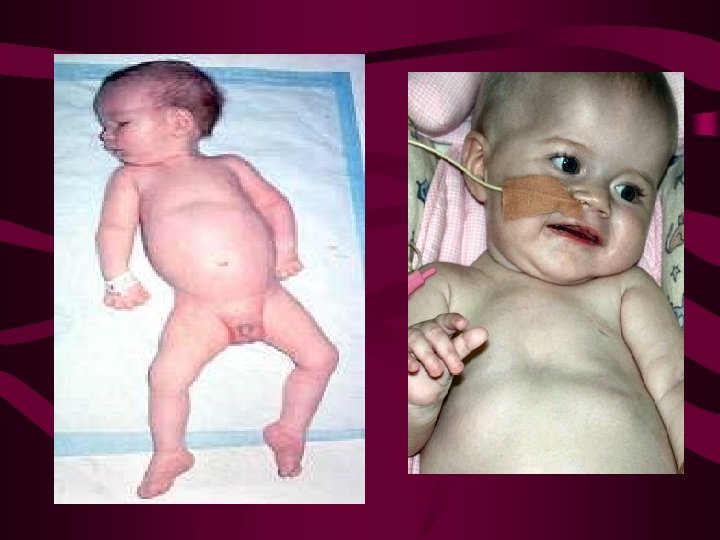
ATROFIA MUSCULAR ESPINAL INF Y JUVENIL • Incidencia: 1 por 6. 000/10. 000 nacimientos • Clasificación: en función de la edad de inicio, AME 1 (Werdnig-Hoffman), AME 2 (intermedia), AME 3 (Kugelberg-Welander) • Genética: los tres tipos mapean en el cromosoma 5, contiene genes SMN 1 y 2 NAIP(proteina inhibidora de la apoptosis neuronal)
Clínica AME 1, AME 2 y AME 3 • AME 1: inicio antes del nacimiento, nunca se pueden sentar y fallecen alrededor de 2 a. por neumonía • AME 2: inicio 18 m. , se pueden dar vueltas y llegan a sentarse, rara vez llegan a caminar, pronóstico varía (inf- 4ª decada) • AME 3: inicio 5 -15 años, debilidad para caminar, posterior/ ms. escapulares y cuello, pseudohipertrofia pantorrillas
Diagnóstico y D. diferencial • • • Dx: demostrar mutación SMN 1 EMG: muestra signos de denervación Biopsia: atrofia de grupo CK: puede estar elevada en AME 3 D. Dif: con miopatías y distrofias ms congénitas AME 1, con distrofias, miopatías metabólicas, infl. y endocr. AME 2 y 3
OTRAS AME • Atrofia espinal del adulto, AME 4: inicio después de los 20 a. , rara vez se encuentra deleción SMN 1, general/ solo afecta cinturas • Atrofia bulbar progresiva de la infancia o enfermedad de Fazio-Londe: paresia facial y bulbar progresiva de la infancia, a veces fallo respiratorio.
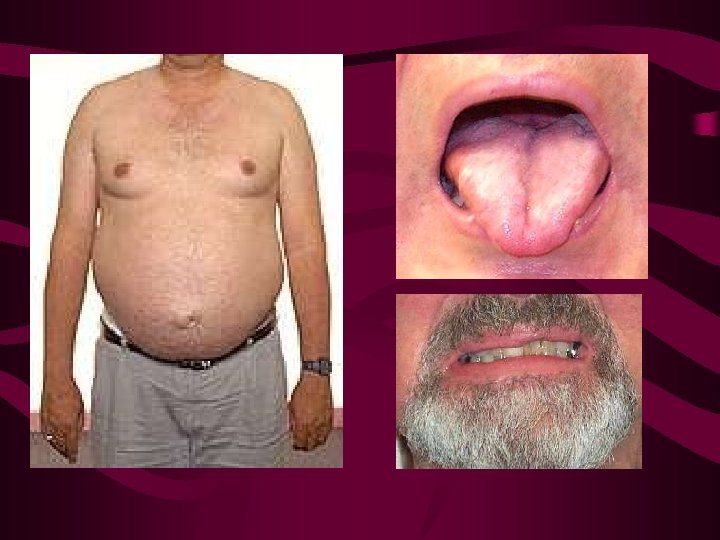
Enf. de Kennedy o atrofia bulboespinal X-L • Inicio alrededor de los 30 a. con debilidad progresiva de los ms. cinturas tipo MNI, debilidad facial y amiotrofia lingual. • Disartria y disfagia en 50% • Calambres/fasciculac > faciales y periorales • Ginecomastia 60 -90% • Otras: atrofia testicular, feminización, disminución de la fecundidad, diabetes mellitus. No espasticidad Puede haber neuropatía sensitiva
Enf. Kennedy o atrofia bulboespinal X-L: Laboratorio • • Alteración EMG (puede haber neurop. Sensitiva) Elevación CK Alteraciones hormonales Genética: expansión del triplete CAG de receptor del gen de los andrógenos localizado en el cromosoma X.
Enfermedad de Tay-Sachs del adulto • Afectación predominante de la MN inferior producida por un déficit de hexosaminidasa beta (hex A) • Se diferencia de la ELA en que tienen una progresión muy lenta y puede haber disartria y atrofia cerebelosa manifiesta en RM
ENFERMEDADES DE LA MNS Y MNI: ELA • Causa indeterminada • Variantes: ELP, AEP, ABP • Dinámica y progresiva: casos de inicio MNS y MNI, a veces inicio MNS o MNI • 5 -10% casos son hereditarios (AD) • Prevalencia 3 -8/100. 000, ratio H/M: 1. 2 -1. 6/1 • Pico enfermedad : 6ª década (2ª-85 a) • Duración media: 3 años (>1 década-meses)
ELA: etiología • • ELA familiar (10% de ELA) Excitotoxinas Estrés oxidativo Disfuncion neurofilamentos Alteración homeostasis del Ca Disfunción mitocondrial Activación apoptosis Citoquinas proinflamatorias
Formas familiares en ELA 1. Mutaciones SOD (superoxido dismutasa) 20% de ELA familiar, 1% de casos esporádicos 2. Senataxina Inicio juvenil Curso lentamente progresivo 3. FUS 4 -5% ELA familiar, 0, 5 -0, 7% de ELA esporádica 4. VAP (3 variantes) -ELA atipica calambres y fasciculaciones de curso lentamente progresivo -ELA rápidamente progresiva -Atrofia muscular espinal de inicio tardio
Formas familiares ELA 5. TDP-43, dominante Curso lentamente progresivo en la mayoría, rápido en algunas familias 6. Angiogenina, dominante Forma clínica típica 7. FIG 4, dominante o esporádico 1 -2% de ELA 8. Dinactina, dominante Inicio ms craneal, solo 1 familia hallada 9. Alsina, recesiva Inicio en infancia, generalmente antes de 10 años. Muy lentamente progresiva 10 y 11. Otras: cadena pesada neurofilamentos, Periferina
ELA: clínica • Debilidad focal que se disemina a ms. contiguos • Inicio más frecuente en extremidades inferiores (ELA clásica), en 25% casos el inicio es bulbar (ELA bulbar) • Rara vez inicio respiratorio (forma disneica) • A veces debilidad hemicuerpo (forma Mills) • Otros: fasciculac. , calambres, fatiga, alteraciones del sueño por hipoxia • No debe de haber: alt. sensib, extrapiramidales, demencia, paresia OCM, esfínter Alterac función ejecutiva: 20 -40% y demencia frontotemporal 5%
ELA: a) Reposo, b) protrusión A B
ELA: diagnóstico • Hª, examen clínico y curso de la enfermedad • Laboratorio obligado: hemograma, bioquímica de sangre que incluya CK, VSG, RPR, hormonas tiroideas, IEF, B 12, Rx tórax. • Si solo afectación MNI: Ac anti. GM 1 • Electrofisiología: i)confirmar alteración MNI en las regiones clínica/ afectadas, ii) confirmar extensión a las clínica/ no afectadas, iii) excluir otros procesos patológicos) descartar bloqueos o desmielinización • RM encefálica y espinal: descartar tumores, cervicoartrosis, otras
RM: hiperintensidad FLAIR
El Dx de ELA requiere: • (A: 1) evidencia de afectación MN inferior (MNI), clínica, electrofisiológica o patológica • (A: 2) evidencia de afectación de MN superior en el examen clínico • (A: 3) diseminación progresiva de los síntomas, de una región a otra, determinada por la Hª o el examen.
ELA: criterios diagnósticos • Junto con la ausencia de: • (B: 1) evidencia electrofiológica o patológica de otra enfermedad que pudiera explicar los signos de degenerac. de MNI o MNS, y • (B: 2) evidencia de otra enfermedad en la neuroimagen que pudiera explicar los signos clínicos y electrofisiológicos observados.
ELA: D. diferencial • • Tumores (RM) Siringomielia, siringobulbia (RM) Mielopatía cervical espondiloartrósica (RM) Neuropatía motora multifocal (EMG) En casos bulbares miastenia gravis Todos los Dx dif de ELP Todos los DX de AMP
ELA: manejo y trto • farmacoterapia específica (Riluzole, acción antiglutamatérgica) • tratamiento sintomático (dietético, del insomnio, depresión, fallo respiratorio, etc) • rehabilitación física (estiramientos, terapia ocupacional, dispositivos ortopédicos de ayuda) • atención a las alteraciones de lenguaje (logopeda) • cuidado nutricional (espesantes, gastrostomía) • cuidado respiratorio (ventilación no invasiva, traqueotomía y ventilación invasiva)
ELA: otras asociaciones • Complejo ELA- Parkinson- Demencia, en la isla de Guam (Pacifico Oeste), en Nueva Guinea y el la Península de Kii (Japon), la incidencia de ELA es entre 50 -150 veces más frecuente que en otros lugares. Se piensa que factores genéticos y ambientales tengan que ver en la patogénesis.